










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Child Health
On-line version ISSN 1999-7671Print version ISSN 1994-3032
S. Afr. j. child health vol.11 n.3 Pretoria Oct. 2017
https://doi.org/10.7196/sajch.2017.v11i2.1312
RESEARCH
Fanconi anaemia in South African patients with Afrikaner ancestry
C FebenI; T HawII; D StonesIII; C JacobsIV; C SuttonV; J KrombergVI; A KrauseVI
IMB BCh, DCH, MMed, FCMG (SA); Division of Human Genetics, National Health Laboratory Service, and Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMSc (Med);Division of Human Genetics, National Health Laboratory Service, and Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIFCPaed (SA); Department of Paediatrics, Universitas Hospital; and University of the Free State, Bloemfontein, South Africa
IVMB MCh; Unitas Hospital, Pretoria, South Africa
VMB MCh, DTM& H, DPH, DCH, FCPaed (SA); Department of Paediatrics, Polokwane Mankweng Hospital Complex, Polokwane and University of Limpopo, Polokwane, South Africa
VIPhD; Division of Human Genetics, National Health Laboratory Service, and Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
BACKGROUND. Fanconi anaemia (FA) is a rare genetic disorder of impaired DNA repair that results in physical and haematological consequences in affected individuals. In South Africa (SA), individuals with Afrikaner ancestry are at an increased risk of inheriting disease-causing FA mutations, owing to the three common FANCA (FA, complementation group A) founder mutations present in this population subgroup.
OBJECTIVES. To describe the physical phenotype of SA patients with FANCA mutations for the purpose of recommending appropriate care for affected individuals.
METHODS. A structured clinical examination and file-based review were used to evaluate the physical phenotype of 7 patients with compound heterozygous and homozygous FANCA founder mutations, and 1 patient with confirmed FANCA complementation analysis. Descriptive statistical analysis was used to determine the frequency of physical anomalies in Afrikaner patients and to compare the described phenotype to other FA cohorts, including a previously clinically characterised black SA FA cohort.
RESULTS. An earlier age of diagnosis of FA in Afrikaner patients, a high frequency of somatic anomalies and a higher-than-expected incidence of the VACTERL/H phenotype were noted.
CONCLUSIONS. Based on our findings, recommendations for the care of FA patients with Afrikaner ancestry are made, including renal ultrasound evaluation at diagnosis and hearing screening.
Fanconi anaemia (FA) is a rare genetic condition of impaired DNA repair mechanisms and chromosomal instability. Although most cases are inherited in an autosomal recessive manner, both autosomal dominant and X-linked recessive patterns of inheritance have been described.[1-3] In general, the FA phenotype is characterised by a broad spectrum of congenital anomalies, principally involving growth, skin pigmentation and dysmorphogenesis of the skeletal, cardiovascular, genito-urinary, gastrointestinal and central nervous systems (CNS).[4] Internationally, major congenital malformations are reported in over two-thirds of patients.[5] Affected individuals are further at risk for childhood-onset haematological disease, which is characterised by initial thrombocytopenia and macrocytosis and progression to aplastic anaemia, acute myeloid leukaemia (AML) or myelodysplastic syndrome (MDS).[5] The incidence of solid tumours, particularly of squamous-cell lineage (including cancers of the head and neck, oesophagus, vulva and cervix), is also significantly increased in affected patients. These tumours are recognised particularly in developed nations, where improved treatments of the haematological complications of the disease, including haematopoietic stem-cell transplantation, are readily available.[5,6]
While FA is a genotypically heterogeneous disorder, caused by mutations in at least 21 different FA genes (FANCA, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P, Q, R, S, U, V),[2,3,7-9] mutations in 3 of these genes account for a high proportion of diagnosed cases - FANCA, 60.5%; FANCC, 16%; and FANCG 10%.[5] Furthermore, certain population groups (e.g. Ashkenazi Jews, Spanish Gypsies, Japanese) have been shown to harbour founder mutations that account for a high percentage of cases in these specific groups.[10-12] In South Africa (SA), founder mutations have been characterised in both the black and caucasian Afrikaans-speaking populations.[13,14]
Black SA patients with FA have been shown to carry a founder mutation in the FANCG gene (c.637_643delTACCGCC -a7 base-pair (bp) deletion mutation).[14] This founder mutation, in the homozygous state, accounts for ~80% of affected black patients, and has resulted in an estimated birth incidence of FA in black South Africans nearing 1/40 000.[14] The physical and haematological phenotypes of black patients, homozygous for the 7 bp deletion mutation, have been characterised previously.[15,16]
In SA individuals with Afrikaner ancestry, three null mutations in FANCA (del E12-31 (deletion mutation of exons 12 to 31); del E11-17 (deletion mutation of exons 11 to 17) and 3398delA (point mutation at position 3398)) have previously been shown to account for ~80% of FA cases.[13] Molecular and genealogical evidence has confirmed that these founder FANCA mutations were probably introduced into SA following the 17th century migration of the French Huguenots to the Cape.[13] Based on birth incidence and point-prevalence data, respectively, the prevalence of FA in individuals with Afrikaner ancestry has previously been estimated to approximate between 1/22 000 and 1/26 000.[17] Despite the predicted relative high prevalence of FA in Afrikaner individuals, there is little published work on the physical and haematological phenotypes of these patients.[17,18] The most comprehensive study was performed in 1994,[18] prior to the introduction of molecular testing methods to characterise specific mutations and therefore no detailed mutation-specific genotype-phenotype correlations could be drawn.
The aims of the present study were firstly, to describe the physical phenotype of affected Afrikaner patients with confirmed founder FANCA mutations, and secondly, to compare this phenotype with the physical phenotype in black South African patients with FA caused by the homozygous FANCG deletion mutations described by Feben et al.[15]and other FA cohorts with mutations in FANCA. An improved recognition of the physical phenotype of FA in patients with Afrikaner ancestry may assist in improving the early recognition and diagnosis of the condition, and subsequently, the care offered to affected patients.
Methods
Patients for this study were recruited from tertiary-level haematology/ oncology clinics in the cities of Pretoria, Bloemfontein and Polokwane, which are situated in 3 of the 9 provinces of SA, between October 2009 and July 2012. Altogether, 8 (self-reported ancestry) Afrikaner patients were recruited, of whom 7 were homozygous or compound heterozygous for the FANCA founder mutations. The 8th patient had positive chromosome-breakage testing with confirmed FANCA complementation analysis, but did not consent to further molecular testing.
Molecular genetic testing, combining polymerase chain reaction (PCR) and multiplex ligation probe specific amplification (MLPA) techniques, is used as the initial testing method in Afrikaner patients suspected to have FA, in view of the high percentage of cases caused by founder mutations in SA (personal communication, Prof. A Krause, January 2016). Molecular genetic testing of the 7 patients was performed by the Molecular Genetics Laboratory at the National Health Laboratory Services in Johannesburg, SA, with assistance from a collaborating laboratory in one case (Sheffield Diagnostic Genetics Services, UK).
A comprehensive clinical examination and a concurrent review of each patient's hospital records were completed. The clinical examination aimed to document growth measurements (current weight, height and head circumference); upper-limb and lower-limb anomalies, with particular focus on abnormalities of the radial ray and hands, and skin-pigmentary abnormalities. All measurements were plotted on relevant growth charts for comparison with the age-related mean. Major malformations of the renal, genital and cardiovascular systems were documented, as well as the results of hearing-screening tests. Information from the patients' hospital records was used to document the age of presentation at the haematology/oncology clinic, and the clinical symptoms at initial presentation.
Descriptive statistical analysis of the data was performed. The median current age and median age of presentation with symptoms in keeping with FA were calculated. The frequency of somatic anomalies and growth disturbances were recorded, and compared with those of other cohorts with documented FANCA mutations, as well as with the Rosendorff et al.[l7]and MacDougall et al.'181 cohorts, which represent the only major study groups comparable with the present cohort. Descriptive comparisons were also made with the previously published data on the physical phenotypic characteristics in black South African patients with FA caused by a homozygous FANCG founder mutation.[15]
A modified International FA Registry (IFAR) score (using growth retardation, renal anomalies, thumb and/or radius anomalies, microphthalmia and birthmarks (each scoring positive 1 if present) and other skeletal anomalies (scoring negative 1 if present, for a maximum score of 5)) was calculated for each patient. The IFAR score was initially described by Auerbach et al.[19] as a screening tool to predict the likelihood of an individual having FA. The original score comprises the above five criteria, as well as low platelet count (score positive 1 if present) and developmental delay (score negative 1 if present), giving a maximum score of 6.[19] An adjusted score was used, as we did not have platelet counts and neurodevelopmental assessments for each patient. Additionally, a modified pigmentation, small head, small eyes, CNS (not hydrocephalus), otology, and short stature (PHENOS) score was calculated for each patient. The PHENOS score, developed by Alter and Giri,[20] allocates a score of positive 1 for each of: pigmentation anomaly, microcephaly (small head), microphthalmia (small eyes), CNS anomalies (other than hydrocephalus), otologic anomalies (structural ear anomalies or hearing loss) and short stature, with a maximum score of 6. The score is used to identify patients with VACTERL/H association (which includes at least three of the following cardinal abnormalities: vertebral, ano-rectal, cardiac, tracheo-oesophageal, renal, limb defects and hydrocephalus), who should be tested for FA.[20] We used a modified score as we did not have CNS imaging in our patients.
The study was approved by the research ethics committees of the University of the Witwatersrand (ref. no. M090681), the University of the Free State (ref. no. ETOVS NR 52/2010) and the University of Limpopo (ref. no. PMREC-19).
Results
The median age of the 8 patients attending the haematology/oncology clinics at the time of the study was 11 years 11 months (range 4 years 1 month - 17 years 3 months). The initial presenting complaints were variable, and included bleeding diatheses (epistaxis, haematemesis), fatigue and somatic abnormalities (particularly involving the thumbs), with the median age at presentation being 3 years (range birth - 12 years).
The physical phenotype of each patient (N=8) was recorded (Fig. 1). The most frequently occurring anomalies were head circumference below the 10th centile for age (75%), with head circumference below the 3rd centile for age in 3 of these patients (37.5%); radial-ray anomalies (87.5%); and skin-pigmentation abnormalities (87.5%). Two patients met the criteria for the diagnosis of VACTERL/H association. The median modified PHENOS score was 2 (range 1 - 3), with the 2 patients who met criteria for VACTERL/H association scoring 3 (patient 2) and 2 (patient 7). The median modified IFAR score was 3 (range 2 - 4). Although this score cannot be directly used to predict the likelihood of having FA, given that we did not calculate the full score (in the absence of haematological data and a neurodevelopmental assessment), the high median value points to the high frequency of congenital malformations in the present cohort. A score of 3 using the complete IFAR score would correlate with a >92% risk of having FA.
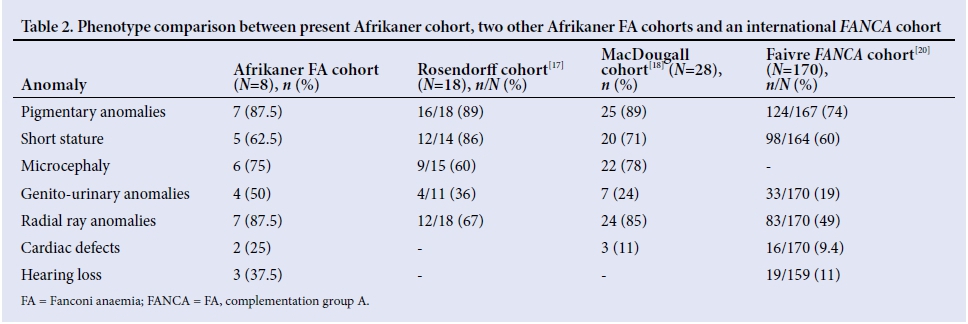
The physical phenotype of the 8 patients in the present cohort was compared with the physical phenotypes of the patients in the Rosendorff et al.[17] (N=18) and MacDougall[18] (N=28)cohorts (Table 1). No significant differences were found in the frequency of anomalies between the groups (using two-tailed Fisher's exact test). Most patients in the Rosendorff et al.[17] cohort had an IFAR score >4, also indicating the high number of somatic anomalies in each patient. Given that we now know the molecular basis of FA in Afrikaner patients, we can assume that at least 80% of the patients in the above 2 cohorts would have carried founder FANCA mutations as the cause of their FA. A comparison to a large (N=170) international FANCA cohort also showed no significant differences in the frequency of all evaluated anomalies (Table 1).[21]
The Afrikaner FA physical phenotype was compared with that previously detailed in black SA FA patients who are homozygous for a FANCG 7 bp deletion founder mutation (N=35) (Table 2).[15] No significant differences were noted between the 2 groups regarding the frequency of the anomalies; however, the sample size of the present cohort was small. The trend appears to indicate that physical anomalies (radial defects, renal malformations) may be more common in Afrikaner FA patients with FANCA mutations, and suggests that analysis of a larger Afrikaner cohort may be beneficial in determining whether this trend is significant. The age of presentation with symptoms or signs suggestive of FA was significantly lower in Afrikaner patients than in black FA patients (median age at presentation 3 years v. 7 years 8 months, p<0.001).[15] Additionally, 3 of the Afrikaner patients presented with a diagnosis of FA before 6 months of age, on the basis of congenital anomalies, whereas none of the black patients presented before 2 years 11 months, suggesting that the physical anomalies in Afrikaner patients were clinically more apparent or more severe in presentation.[15] This is further evidenced by the differences in the description of the radial anomalies in the two groups. The radial-ray anomalies noted in black patients were described as subtle, with none of the patients showing radius hypoplasia or aplasia, whereas the Afrikaner patients had very easily identifiable radius and radial-ray defects, 2 cases having bilaterally absent thumbs.[15]
Discussion
FANCA mutations account for the highest percentage of FA patients worldwide as documented by the IFAR. Numerous common and family-specific deleterious mutations have been reported in this gene, which appears to function as part of the core nuclear complex in the FA pathway.[22] Previous research efforts have variably focused on complementation-specific and mutation-specific genotype-phenotype correlations in order to better elucidate the physical and haematological consequences of mutations in FANCA. Clinical phenotyping aims to expedite diagnosis and improve care and management of affected individuals.
Two large international studies characterised the clinical phenotype of individuals with different mutations in the FANCA gene. The initial study carried out in 2000 found that FANCA individuals with homozygous null mutations had a higher frequency of somatic anomalies than individuals with FANCC mutations, but a similar frequency to those with FANCG mutations. FANCA individuals were also at increased risk for severe haematological disease. A number of the patients in this study were of Afrikaner ancestry and had at least one null del12-31 mutation.[21] In 2011, research into the functional role and clinical impact of FANCA mutations challenged the earlier findings. They demonstrated no correlation between mutation type and physical or haematological outcome, indicating that mutation-specific genotype-phenotype correlations may not be useful in individuals with FANCA mutations. The authors concluded that mutation type had little prognostic value in FANCA patients and that other factors, including genetic background, ethnicity and environmental exposure were more important in determining the clinical outcome.[12] Despite the different conclusions drawn in these two studies, they both support the importance of clinical phenotyping in individuals with FANCA mutations. More specifically, the later study suggested that characterisation of the FA phenotype based on ethnicity may be as important as mutation-specific phenotype correlations, and that population-specific phenotypes may exist independently of the specific mutation/s.[12] A similar focus on the underlying importance of ethnicity was highlighted by Futaki et al.ln] The study showed that a specific mutation (IVS4+4A>T) in the FANCC gene caused a significantly milder clinical phenotype in Japanese patients than in Ashkenazi Jewish patients.
Previous SA research studies documented the physical phenotype of Afrikaner patients with FA based on home language and self-reporting; however, at this point molecular pathogenesis of the condition had not been characterised.[17'18] It is now known that the majority of Afrikaner patients with FA harbour founder FANCA mutations as the cause of their condition.
The results of the present study confirm the high number of somatic anomalies found in Afrikaner patients with FA due to founder mutations. They also show that Afrikaner patients are diagnosed at a significantly younger age than black patients in SA. While this may partially reflect social factors like access to tertiary healthcare' it is important to note that the somatic anomalies in Afrikaner patients are often recognisable and obvious at birth' and that these anomalies should prompt consideration of an FA diagnosis and further investigations.[15]
The results support the use of the IFAR scoring system as an important screening tool for FA in Afrikaner patients. The use of screening tools is particularly important in countries, such as SA, where access to genetic services and particularly molecular genetic testing is limited by poor resources and funding. Once an individual is suspected of having FA, the results in the present cohort suggest that these individuals, at least, should be offered renal ultrasound screening and hearing tests as part of their care.
Two patients met the clinical criteria for a diagnosis of VACTERL/H association. Previous reports differed vastly in their reporting of the percentage of patients with FANCA mutations who meet the diagnostic criteria for VACTERL/H association, with figures of between 3% and 20%.[20,24] Although the VACTERL/H phenotype appears to be more often associated with mutations in FANCD1 (33%), E (40%) and F (30%), more recent work by Alter and Giri´[20] suggests that the co-occurrence of this phenotype with FA is under-recognised.[24] The present sample size is however small and the results suggest that Afrikaner patients with a VACTERL/H phenotype should always be tested for FA. Future studies with a larger cohort will allow a more accurate frequency measurement of the VACTERL/H phenotype in this population subgroup. Further, the small sample size questions the viability of the PHENOS score in Afrikaner patients. Even so, this score may be useful in stratifying patients with the VACTERL/H phenotype for FA testing in future studies.
Despite the high frequency of FA and high incidence of congenital anomalies in the Afrikaner population subgroup, anecdotal evidence suggests that very few affected patients ever attend a genetic clinic or have their clinical diagnosis confirmed with a molecular test in SA. Certainly, in our study, we were only able to ascertain 7 patients with molecular confirmation of their condition despite accessing three tertiary healthcare centres in three cities in SA. The reason for this under-recognition and under-diagnosis of FA in the Afrikaner population in SA remains unclear. It is concerning that the patient referral rate to genetic services, tertiary haematology and oncology services is so low. It is possible that a more severe end of the spectrum exists, with affected individuals dying at a young age, before consideration of the diagnosis of FA (possible misdiagnosis of isolated VACTERL/H association); or that a much milder undiagnosed phenotype exists with few congenital anomalies and limited haematological complications. An area for future research includes a molecular screen for FANCA mutations in Afrikaner patients with VACTERL/H association.

Conclusion
The results indicate that Afrikaner patients with FA have a high incidence of congenital anomalies and yet very few have their diagnoses confirmed on a molecular level. As such, we recommend that all children with one or more significant or major congenital anomaly (a birth defect that may cause death or disability) be referred for a tertiary evaluation by a paediatrician or medical geneticist. The diagnosis of FA should be considered on the basis of the congenital malformation, especially in the presence of other clinical features (particularly skin pigmentary anomalies) to diagnose FA prior to the onset of haematological disease. In the SA setting, molecular FANCA analysis of the three founder mutations would be the first line of investigation to confirm the diagnosis in an individual with Afrikaner heritage. Through earlier diagnoses, we can improve care and surveillance, provision of genetic counselling (including recurrence risk counselling) and provide an opportunity for the affected patient to be considered for haematopoietic stem cell transplantation.
Acknowledgements. The authors wish to thank the clinical, counselling, laboratory and support staff in the Division of Human Genetics (National Health Laboratory Service and The University of the Witwatersrand), clinical and support staff at Universitas Hospital, Polokwane/Mankweng Hospital Complex and Unitas Hospital and the patients and their families who agreed to participate in this research. We thank our partners at the Sheffield Regional Genetics Laboratory for their assistance.
Author contributions. Study design and conception: CF, AK, TH, JK; data collection and analysis: CF, AK, TH, JK, DS, CJ, CS; clinical collaboration: DS, CJ, CS; writing of article: CF, JK, AK; editing: CF, JK
Funding. Medical Research Council of South Africa.
Conflicts of interest. None.
References
1. Meetei AR, Levitus M, Xue Y, et al. X-linked inheritance of Fanconi anaemia complementation group B. Nat Genet 2004;36:1219-1224. https://doi.org/10.1038/ng1458 [ Links ]
2. Alter B, Kupfer G. Gene Reviews: Fanconi Anemia. Bethesda: National Center for Biotechnology Information, 2013. https://www.ncbi.nlm.nih.gov/books/NBK1401/ (accessed 15 July 2016). [ Links ]
3. Ameziane N, May P, Haitjema A, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun 2015;6:8829. https://doi.org/10.1038/ncomms9829 [ Links ]
4. Shimamura A, Alter B. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 2010;24(3)101-122. https://doi.org/10.1016/j.blre.2010.03.002 [ Links ]
5. Auerbach A. Fanconi anaemia and its diagnosis. Mut Res 2009;668(1-2):4-10. https://doi.org/10.1016/j.mrfmmm.2009.01.013 [ Links ]
6. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anaemia. Blood 2003;101(3):822-826. https://doi.org/10.1182/blood-2002-05-1498 [ Links ]
7. Sawyer SL, Tian L, Kahkonen M, et al. Biallelic mutations in BRCA1 cause a new Fanconi anaemia subtype. Cancer Discoveries 2015;5(2):135-142. https://doi.org/10.1158/2159-8290.cd-14-1156 [ Links ]
8. Park JY, Virts EL, Jankowska A, et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet 2016;53(10):672-680. https://doi.org/10.1136/jmedgenet-2016-103847 [ Links ]
9. Bluteau D, Masliah-Planchon J, Clairmont C, et al. Biallelic inactivation of REV7 is associated with Fanconi anaemia. J Clin Invest 2016;126(9):3580-3584. https://doi.org/10.1172/jci88010 [ Links ]
10. Whitney MA, Saito H, Jakobs PM, Gibson RA, Moses RE, Grompe M. A common mutation in the FACC gene causes Fanconi anaemia in Ashkenazi Jews. Nat Genet 1993;4(2):202-205. https://doi.org/10.1038/ng0693-202 [ Links ]
11. Yagasaki H, Oda T, Adachi D, et al. Two common founder mutations of the fanconi anaemia group G gene FANCG/XRCC9 in the Japanese population. Hum Mut 2003;21(5):555. https://doi.org/10.1002/humu.9142 [ Links ]
12. Castella M, Pujol R, Callen E, et al. Origin, functional role and clinical impact of Fanconi anaemia FANCA mutations. Blood 2011;117(14):3759-3769. https://doi.org/10.1182/blood-2010-08-299917 [ Links ]
13. Tipping AJ, Pearson T, Morgan NV, et al. Molecular and genealogical evidence for a founder effect in Fanconi anaemia families of the Afrikaner population of South Africa. Proc Natl Acad Sci USA 1993;98(10):5734-5739. https://doi.org/10.1073/pnas.091402398 [ Links ]
14. Morgan NV, Essop F, Demuth I, et al. A common Fanconi anaemia mutation in black populations of sub-Saharan Africa. Blood 2005;105(9):3542-3544. https://doi.org/10.1182/blood-2004-10-3968 [ Links ]
15. Feben C, Kromberg J, Wainwright R, et al. Phenotypic consequences in black South African Fanconi anaemia patients homozygous for a founder mutation. Genet Med 2014;16(5):400-406. https://doi.org/10.1038/gim.2013.159 [ Links ]
16. Feben C, Kromberg J, Wainwright R, et al. Haematological consequences of a FANCG founder mutation in black South African Patients with Fanconi anaemia. Blood Cells Mol Dis 2015;54(3):270-274. https://doi.org/10.1016/j.bcmd.2014.11.011 [ Links ]
17. Rosendorff J, Bernstein R, MacDougall L, Jenkins T, Opitz JM, Reynolds JF. Fanconi anaemia: Another disease of unusually high prevalence in the Afrikaans population of South Africa. Am J Med Genet 1987;27(4):793-797. https://doi.org/10.1002/ajmg.1320270408 [ Links ]
18. MacDougall LG, Rosendorff J, Poole J, Cohn RJ, McElligott SE. Comparative study of Fanconi anaemia in children of different ethnic origin in South Africa. Am J Med Genet 1994;52(3):279-284. https://doi.org/10.1002/ajmg.1320520306 [ Links ]
19. Auerbach AD, Rogatko A, Schroeder-Kurth TM. International Fanconi Anaemia Registry: Relation of clinical symptoms to diepoxybutane sensitivity. Blood 1989;73:391-396. [ Links ]
20. Alter B, Giri N. Thinking of VACTERL-H? Rule out Fanconi anaemia according to PHENOS. Am J Med Genet 2016;170(6):1520-1524. https://doi.org/10.1002/ajmg.a.37637 [ Links ]
21. Faivre L, Guardiola P, Lewis C, et al. Association of complementation group and mutation type with clinical outcome in Fanconi anaemia. Blood 2000;96:4064-4070. [ Links ]
22. Levran O, Diotti R, Pujara K, Batish SD, Hanenberg H, Auerbach AD. Spectrum of sequence variations in the FANCA gene: An International Fanconi Anaemia Registry (IFAR) study. Hum Mut 2005;25(2):142-149. https://doi.org/10.1002/humu.20125 [ Links ]
23. Futaki M, Yamashita T, Yagasaki H, et al. The IVS4+4A>T mutation of the Fanconi anaemia gene FANCC is not associated with a severe phenotype in Japanese patients. Blood 2000;95:1493-1498. [ Links ]
24. Faivre L, Portnoi MF, Pals G, et al. Should chromosome breakage studies be performed in patients with VACTERL association? Am J Med Genet A 2005;137:55-58. https://doi.org/10.1002/ajmg.a.30853 [ Links ]
 Correspondence:
Correspondence:
C Feben
candice.feben@nhls.ac.za

{kind=link}
{kind=link}