










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSAMJ: South African Medical Journal
On-line version ISSN 2078-5135
Print version ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.108 n.5 Pretoria May. 2018
http://dx.doi.org/10.7196/samj.2018.v108i5.13004
REVIEW
Fanconi anaemia in South Africa: Past, present and future
C FebenI; T WainsteinII; J KrombergIII; F EssopIV; A KrauseV
IFCMG (SA); Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIMSc Med (Genetic Counselling); Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIPhD;Division of Human Genetics, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IVMSc; Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
VPhD; Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
Fanconi anaemia (FA) is an inherited genetic disorder characterised by somatic anomalies, bone marrow failure and an increased predisposition to solid tumours and haematological malignancies. South African (SA) black and Afrikaner individuals are at higher than average risk for this condition owing to genetic founder mutations in certain Fanconi-associated genes. This review explores the epidemiology, clinical presentation, diagnostic modalities and recommended care of affected patients, focusing on the founder population groups in SA. The early diagnosis of FA is important and provides improved opportunities for early intervention, but remains challenging.
Fanconi anaemia (FA) is an inherited disorder of impaired DNA repair, first described by Swiss paediatrician Guido Fanconi in 1927 and since then extensively documented in individuals of diverse ethnic origin.[1-3] Characterised clinically by somatic, haematological and oncological anomalies, most often manifesting in childhood, the condition is cytogenetically typified by spontaneous and induced chromosome breakage and is usually inherited in an autosomal recessive manner.[1] In South Africa (SA), in contrast to other populations worldwide where genotypic heterogeneity typically underpins this complicated disorder,[1] two population subgroups with well-characterised founder mutations have allowed for focused and genotype-specific research into FA.[4-6] However, despite our research and enhanced understanding of this disorder, the diagnosis of FA remains a clinical and laboratory challenge in the SA healthcare system.
This article reviews the epidemiology, molecular pathogenesis, clinical phenotype, available local and international genetic testing and recommended surveillance and treatment for FA, with a focus on the local black and Afrikaner population groups. Additionally, we discuss some rare cases of FA caused by mutations in non-founder FA genes that have highlighted a need for broadened genetic testing capacity for this condition in SA.
The term 'black' is used to describe individuals of indigenous ancestry originally descended from sub-Saharan Bantu-speaking groups and in whom genetic founder mutations have been noted. The term 'Afrikaner' is used to describe Caucasian individuals of self-reported Dutch-European descent.
Epidemiology
FA has been reported in numerous racial and ethnic groups and is considered worldwide to be an important heritable cause of aplastic anaemia in children.[7] The condition is usually inherited in an autosomal recessive manner, although autosomal dominant and X-linked forms are described, with a slight male preponderance (skewing of the male-to-female ratio 1.2:1 v. expected 1:1). North American carrier frequencies are estimated at 1/181. The carrier rate and the expected prevalence of the condition in two subgroups of the SA population (as well as other population subgroups elsewhere, including the Ashkenazi Jewish population, the Spanish gypsies and the Japanese) is estimated to be much higher than the quoted North American figure as a result of founder effects.[1] The National Cancer Institute Dictionary of Genetic Terms defines a founder mutation as 'a genetic alteration observed with high frequency in a group that is or was geographically or culturally isolated, in which one or more of the ancestors was a carrier of the altered gene'.[8]
Based on haplotype analysis and evaluation of population genetic data, a founder mutation in the black population (FANCG: NM_0046 29.1g.35077270_35077264del) appears to predate the arrival of Bantu speakers in southern Africa in AD 400, and is therefore an ancient mutation, possibly with its origins in West Africa.[2,5] The estimated carrier frequency of the mutation is 1/100, with the predicted birth incidence of FA in black South Africans approximating 1/40 000.[2] Molecular and genealogical evidence indicates that the three Afrikaner founder mutations (FANCA: NM_000135.2 g.89858955_89818546del; NM_000135.2 g.89847979_89861587del; NM_000135.2 g.89813249_89813249del) were probably introduced into SA following the 17th-century migration of the French Huguenots to the Cape.[3,6] The prevalence of FA in individuals of Afrikaner ancestry, based on birth incidence and point prevalence data, respectively, has been estimated as between 1/22 000 and 1/26 000.[3,6] These figures are at least double those seen elsewhere, despite significant under-ascertainment of FA locally.
Based on these predicted frequencies and the well-documented and reported clinical phenotype, we would anticipate that FA is readily diagnosed in secondary and tertiary healthcare facilities. However, a large discrepancy exists between the number of observed and expected cases of FA and indicates that many cases are unrecognised or undiagnosed. Unpublished data suggest that only one of every 15 cases of FA will have molecular confirmation of the diagnosis in SA (A Krause, personal communication, May 2017). This does not appear to be a situation unique to SA, as it has been widely reported that the diagnosis is most often made at the time of bone marrow failure[9] despite the frequency of associated physical congenital anomalies.
Molecular pathogenesis
The complex FA pathway has been extensively investigated over many years to elucidate the mechanisms by which multiple protein products interact synergistically to provide genome stability and ensure cellular survival through DNA repair mechanisms.[10-12] As of 2017, the FA multistep, S-phase specific cellular response to damaged DNA involved 22 genes, designated FANC A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P, Q, R, S, T, U, V and W.[U0,13-16] Collectively, mutations in FANCA, FANCC and FANCG account for >90% of cases.[1] The pathway is activated when DNA replication is stalled[12-and its critical functions include homologous recombination for inter-strand cross-link repair, nucleolytic incision and translesion DNA synthesis.[11,17] The more recent discovery that BRCA1 (FANCU) and BRCA2 (FANCD1), implicated in the causation of hereditary breast and ovarian cancer syndrome (HBOC), are also part of the FA pathway highlights the interplay between abnormal DNA repair mechanisms and the evolution of cancer.[11,18] Most of the FA genes exhibit mutational or allelic heterogeneity, which is evidenced by the large number of sequence variants and mutations that have been described, particularly in the FANCA and FANCG genes.[19-21]
In >80% of black patients with FA, a homozygous seven base-pair deletion mutation in the FANCG gene (NM_004629.1 g.35077270_35077264del p.Tyr213Lysfs)[2] has been confirmed as the cause of the disease. In a further 5% of cases, black patients with FA are heterozygous for the founder mutation, and preliminary molecular analysis of these FANCG heterozygotes revealed a second FANCG mutation on the other allele in 57% of a small cohort.[22] The molecular aetiology for the so-called 'G-negative' black patients with a clinical and/or cytogenetic diagnosis of FA is poorly understood, although most recently mutations in rarer FA genes, including BRCA2, have been identified.[18]
In SA individuals with Afrikaner ancestry, three mutations in FANCA have been shown to account for ~80% of FA cases.[3]
A founder mutation described in individuals of Ashkenazi Jewish ancestry is also found in SA individuals of this origin. This splice-site mutation in the FANCC gene (IVS4+4A>T) accounts for most cases of FA in Ashkenazi Jewish cohorts worldwide.[23]
There is no published literature on the molecular pathogenesis of FA in SA population groups other than those in which founder mutations exist.
Clinical phenotype
Clinically, FA is characterised by a triad of somatic, haematological and oncological abnormalities. Congenital abnormalities, including skeletal, cardiovascular, genitourinary, gastrointestinal and central nervous system malformations, are well documented,[1,24] as are poor postnatal growth, short stature, pigmentary anomalies and endocrine dysregulation.[25] Internationally, data from large patient cohorts have been collated under the auspices of the International Fanconi Anaemia Registry (IFAR) and composite frequency figures for the physical anomalies observed in genetically heterogeneous cohorts have been documented (Table 1).[1,26]
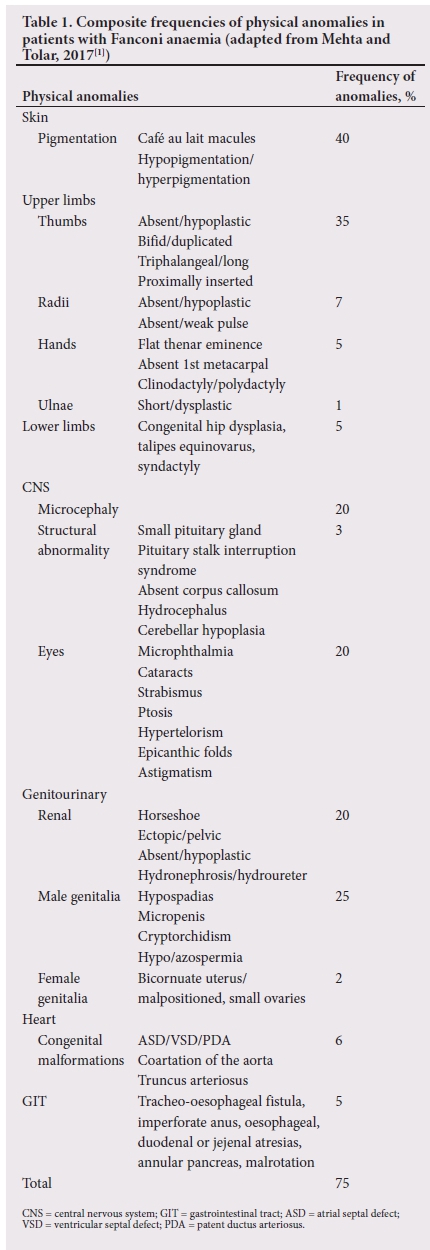
Genotype-phenotype correlation studies performed in the black and Afrikaner populations in SA show that the frequencies of physical anomalies mirror the IFAR data in some respects, but also highlight population- and possibly mutation-specific features in our patient cohorts (Table 2).[4-6] A trend towards significant physical differences between the two founder populations in SA has also been noted and suggests that future research is needed to provide greater definition, particularly for the Afrikaner phenotype.[6] It has been observed that in both population groups, identification and recognition of the FA phenotype may be improved by referral of children with growth restriction, pigmentary anomalies and unusual hands to a paediatrician or medical geneticist for an assessment.[4,6] Certainly in a small Afrikaner patient cohort, the frequency of major congenital anomalies appears high, and patients may meet criteria for the diagnosis of VACTERL-H association ( vertebral, ano-rectal, cardiac, tracheo-esophageal, renal and limb anomalies and hydrocephalus). Additional screening of patients with these abnormalities would be warranted.[6]
From a haematological perspective, bone marrow failure remains the significant identified cause of morbidity and mortality in FA patients, with initial pancytopenia and progression to acute myeloid leukaemia (AML), myelodysplastic syndrome (MDS) or aplastic anaemia.[1,2]
Thrombocytopenia, macrocytosis (raised mean corpuscular volume) and a raised fetal haemoglobin (Hb) usually precede the onset of more severe haematological anomalies.[1,28] Based on data collected by the IFAR, haematological abnormalities appear to present in childhood at a median age of 7 years, with progression to bone marrow failure by the age of 40 years in >90% of patients.[28-Research in a black SA FA cohort determined that the median age of presentation with symptoms of FA, usually concurrent with the diagnosis of bone marrow aplasia, was 7 years and 1 month of age. In the same cohort it was shown that the most common presenting symptom was recurrent epistaxis and that apart from macrocytosis, a severely low Hb (<8 g/dL) was a common initial haematological finding.[5] Based on the ascertainment of patients from haematology/oncology clinics, it is possible that referral bias in this study led to exclusion of more or less severely affected individuals.[5]
FA is also recognised as a cancer susceptibility syndrome.[1,28,29] A predisposition to solid tumours of the head and neck, liver, oesophagus and female genital tract is well described and has become more evident in developed-world countries as the treatment for the haematological complications of the disease has improved.[30] In developed nations, the availability of haematopoietic stem cell transplantation (HSCT) as treatment for bone marrow failure has prolonged life and is therefore predicted to result in a higher prevalence of solid tumours as a common clinical manifestation in later life.[28,29] In SA, solid tumours are rarely seen in patients with FA as most still die in childhood and adolescence from bone marrow disease. HSCT is only available to a minority of patients in the private healthcare sector.[5]
The clinical phenotype of individuals with biallelic BRCA2 mutations as the cause of their FA is characteristically different to that described in more typical cases of FA, with more severe somatic anomalies, earlier-onset haematological compromise and childhood susceptibility to solid tumours, including Wilms tumours and medulloblastomas.[31-34] A case report on two black SA children with FA in whom biallelic BRCA2 mutations were identified (Table 3) highlights the difficulties in care and management of these families, in that the children had severe anomalies associated with their FA phenotype, while the parents are obligate heterozygote carriers of a BRCA2 fault and receive an obligatory diagnosis of HBOC.[18] HBOC is associated with variable risks for breast, ovarian, prostate and pancreatic cancers as well as melanomas.[34] Solid-tumour risk for carriers of other rare FA genes, including FANCN (PALB2) and FANCO (RAD51C), is established in cohorts of patients with breast and/or ovarian cancer, with increased risks for breast cancer (33 - 58% in PALB2)[35] and ovarian cancer (<9% in RAD51C).[36] Whether the mutation spectrum related to tumour risk in heterozygous carriers of mutations in these genes is the same as that seen in carriers of FA is uncertain, although consideration of these potential risks is important. There does not appear to be a significantly increased risk of cancer in carriers of mutations in the more common FA genes, including FANCA, FANCC and FANCG,[37]but larger studies are required to clarify this.
Diagnosis
Traditionally, and possibly still regarded as the gold standard, the diagnosis of FA is confirmed by evaluation of the response of metaphase and interphase cells exposed to diepoxybutane or mitomycin C, two commonly used clastogenic DNA cross-linking agents. FA cells show hypersensitivity to these agents, with increased chromosome breakage and the formation of multiple abnormal structures including multi-radials, tri-radials and breaks.[1,12,38] The results should be compared with normal and positive control cells for standardisation.[1] Chromosome breakage abnormalities are not unique to FA and do not elucidate the underlying molecular abnormality, but rather provide cytogenetic evidence of the disease process.[1]
Diagnostic molecular testing for FA has evolved from predominantly single-gene analysis based on the results of complementation analysis (a cell-based technique used to identify the most likely affected gene) to the use of multi-gene next-generation sequencing (NGS) panels that simultaneously evaluate all known FA genes on a single testing platform. This testing strategy has the advantage of allowing many genes to be tested at the same time, which limits waiting periods involved with sequential testing and, as the costs are falling, may possibly replace the use of chromosome breakage and single-gene analysis for FA diagnostics.[39] A number of commercially available NGS panel tests are available through international laboratories.
In SA, as founder mutations exist in certain population groups, diagnostic mutation-specific testing is currently offered by the Molecular Genetics Laboratory of the Division of Human Genetics, National Health Laboratory Services and School of Pathology (University of the Witwatersrand) in Johannesburg. Testing is offered for the common founder Ashkenazi Jewish mutation in the FANCC gene, for three founder mutations in the FANCA gene in the Afrikaner population, and for the seven base-pair deletion mutation in the FANCG gene in black SA patients (F Essop, personal communication, May 2017). BRCA2 mutation analysis is available through the Department of Human Genetics at the University of the Free State in Bloemfontein for those patients in whom a more severe phenotype is noted that may suggest the FANCD1 (BRCA2) subgroup. Patients with a clinical or haematological phenotype suggestive of FA in whom founder mutation testing is inappropriate or negative are currently unable to access NGS testing in the SA state healthcare system. In this regard, a number of research partnerships have been established to attempt to determine the molecular basis of FA in these individuals. Although the results have not been verified in a diagnostic setting, a number of plausible results, indicating possible deleterious mutations in rare FA genes, may suggest that NGS panel testing should augment the current testing strategy for those who have negative founder mutation analysis, or be used as first-line testing in those who do not have Afrikaner or black ancestry. Such testing is being developed and optimised for SA.
Management strategies
Most patients with FA will be cared for by a multidisciplinary team of specialists with the paediatric haematologist/oncologist and ideally the medical geneticist or genetic counsellor at the forefront, and including other specialist paediatricians depending on the congenital anomalies identified.[1] The aims of care are to ensure correct medical and surgical management of the patient and to provide the patient and the family with genetic counselling and psychosocial support. [40] Treatment protocols will differ according to local expertise and resources. Currently HSCT remains the only available cure for the haematological complications of FA, including MDS and AML. The timing of transplantation is critical and it has been suggested that transplantation be performed before the onset of significant haematological complications, and specifically before the patient requires a number of blood product transfusions.[41] After successful transplantation, affected individuals remain at risk for the development of solid tumours, specifically squamous cell malignancies of the head and neck and female genital tract.[1-28] These systems require regular surveillance to ensure early detection. Treatment is challenging owing to the increased toxicity of chemotherapy and radiation in individuals with FA.[1] Unfortunately HSCT is not routinely available in the state healthcare system in SA, so non-curative palliative measures are often utilised.
In the local setting these non-curative measures include androgen administration, which is the mainstay of initial treatment for FA once marrow aplasia is evident. This therapy has been shown to be effective in up to 50% of individuals within 1 month of commencement. It acts to increase the red cell count and Hb level, with a variable platelet and white cell response.[1] A number of complications have been noted, including liver toxicity and risk of the development of hepatic tumours,[1] hirsutism, acne, hyperactivity, and restricted growth leading to short stature.[1,42] Overall, however, survival is thought to be better in androgen-treated v. non-treated patients.[43] Androgen therapy is not advocated in the pre-anaemic phase of the condition (Hb >8 g/dL) owing to the side-effect profile of the medication.[1] Transfusion of platelets and packed cells is used as supportive therapy during crisis periods and at later stages of the disease for palliation.[1]
A comprehensive list of investigations and care recommendations has been made available by the Fanconi Anaemia Research Fund (FARF).[44] These extensive guidelines indicate the optimal care recommended for patients and families with FA. The guidelines detail both childhood and adult surveillance for the anticipated complications of the condition. Some of these recommendations may be challenging to implement in resource-restricted settings. To streamline the necessary and prudent investigations required in a local context, recommendations for management have been made for both black and Afrikaner patients with FA based on the frequency of certain anomalies in genotype-phenotype correlation studies. These recommendations include a baseline renal ultrasound scan and a hearing test in all diagnosed patients. Additionally, recommendations were made to primary and secondary healthcare practitioners to improve the recognition of FA and refer patients for appropriate paediatric or medical genetic assessment (Fig. 1).[4-6] Studies evaluating the endocrine profile of a cohort of black individuals with FA to assess their need for endocrine screening are currently underway.
SA data on cohorts of affected adult patients with FA are currently lacking. With improvements in therapy and treatment modalities and the concomitant increase in longevity of patients, we may need to determine suitable local recommendations for adolescent and adult cancer screening. Of note, the FARF recommends screening for oral cancers (which affect 14% of FA patients) from the age of 10 years, with 6-monthly clinical assessment by a dentist with knowledge of and experience in detecting pre-malignant lesions of the oral mucosa.[44] Lifestyle advice on the avoidance of alcohol and smoking should also be provided.[44] These recommendations should be considered for our patients, even in resource-restricted settings.
Genetic counselling
Individuals diagnosed with FA and their parents, siblings and other at-risk relatives can all benefit from genetic counselling. The aim of genetic counselling is to provide information and support in the understanding of the condition from medical, genetic and psychosocial viewpoints. In most cases, as FA is inherited in an autosomal recessive manner, the parents of an affected child have a 25% chance of having another affected child. Prenatal and preimplantation genetic diagnosis (PGD) can be offered to the parents of an affected child,[45] although in many cases the diagnosis of FA is made in the first child only after the birth of a second or third child. Prior knowledge of an affected fetus may allow for the option of termination of pregnancy in cases where this is acceptable to the parents, or allow parents to prepare for the birth of an affected child. Ideally, knowledge of the impending birth of an affected child may allow for pre-emptive preparation for HSCT. Prenatal testing can be performed by chorionic villus sampling (at 11 - 14 weeks' gestation) or by amniocentesis (at 16 - 20 weeks' gestation) to obtain fetal cells for assessment. Chromosome breakage analysis is theoretically possible on fetal cells and can be used in the event that the causative mutation in the affected child is unknown.[1] However, molecular targeted mutational analysis is the best option for cases in which mutations have been detected. PGD, which utilises an in vitro fertilisation technique and allows for single-cell testing of an eight- to 16-cell embryo, can also be used to 'select' unaffected embryos for implantation. Additionally, embryos have been successfully tested for 'negative' FA status and 'positive' HLA matching to facilitate HSCT in the affected child.[1] This procedure is very costly and not currently available in the state healthcare system in SA.
In instances where the carrier parents of an affected child are identified as having an increased susceptibility to developing cancers,[37] screening and prophylactic management options can be implemented to mitigate these risks. Risk evaluation as well as screening and management guidelines for HBOC should be guided by clinical judgement, family history and genetic testing results.[45,46] Referral of such FA carriers to a specialist genetic clinic for discussion should be encouraged. Given the variability and complexity of the FA clinical presentation, siblings of individuals with FA would also benefit from evaluation, including genetic testing, to exclude a possible FA diagnosis, which would allow for earlier implementation of the appropriate care. Carrier testing for at-risk relatives (such as the parents' siblings) should also be encouraged; this is especially true in the context of founder populations, in which the carrier frequency of founder mutations is increased.[45]
The psychosocial consequences of a diagnosis of FA in a child have been described as similar to those in childhood cancer diagnoses[47] A constant sense of uncertainty and the stress of lifelong medical care necessities, as well as lack of awareness of FA both in the public domain and among healthcare professionals, constitute some of the ongoing stressors and emotional challenges that parents of children with FA must face.[47] Genetics professionals are in a good position to assist such families, as they are equipped to deal with the complicated technological, medical and genetic information as well as these psychosocial aspects for all family members and throughout the lifespan of the affected child and adult.
Conclusions
This review emphasises important aspects related to the recognition, diagnosis, management, care and prevention of FA, a rare but important inherited disorder in the SA context. Research in our local SA populations over the past several decades has brought to light two ethnic groups (black and Afrikaner populations) that have higher than average carrier rates owing to the presence of founder mutations. This information has been essential as the background to the offering of a diagnostic genetic testing service for FA and provision of genetic counselling services for families of affected individuals. More recent research endeavours have shown that NGS technologies may hold the answers for an improved and fully comprehensive service for all SA patients suspected of having FA. It is hoped that ongoing research initiatives on the clinical phenotype of black and Afrikaner FA patients will allow for enhanced recognition of the diagnosis among healthcare professionals. In the local context, the diagnosis of FA should be considered more readily when characteristic features are present. Like many other inherited disorders, FA is complicated in every aspect and requires management and co-ordination by a multidisciplinary team of healthcare professionals and support structures.
Acknowledgements. We thank the clinical, laboratory, counselling and support staff at the Division of Human Genetics, National Health Laboratory Services and the University of the Witwatersrand, as well as our clinical and laboratory collaborators at paediatric haematology/ oncology clinics at Chris Hani Baragwanath Hospital and Charlotte Maxeke Johannesburg Academic Hospital, Universitas Hospital (and the University of the Free State), the Polokwane/Mankweng Hospital Complex (and the University of Limpopo), and Unitas Hospital (and the University of Pretoria). We also thank our patients and their families for helping us to share their stories.
Author contributions. Study design and conception: CF, AK, JK; data collection and analysis: CF, AK, TW, FE; writing of article: CF, TW; editing: AK, JK.
Funding. Medical Research Council of South Africa, Phyllis Knocker Bradlow Award (Colleges of Medicine of South Africa).
Conflicts of interest. None.
References
1. Mehta PA, Tolar J. Fanconi anaemia. Gene Reviews. Last updated February 2017. http://www.ncbi.nlm.nib.gov/books/NBK1401 (accessed 15 July 2017, 16 October 2017). [ Links ]
2. Morgan NV, Essop F, Demuth I, et al A common Fanconi anaemia mutation in black populations of sub-Saharan Africa. Blood 2005;10(9):3542-3544. https://doi.org/10.1182/blood-2004-10-3968 [ Links ]
3. Tipping AJ, Pearson T, Morgan NV, et al Molecular and genealogical evidence for a founder effect in Fanconi anaemia families of the Afrikaner population of South Africa. Proc Natl Acad Sci USA 2001;98(20):5734-5739. https://doi.org/10.1073/pnas.091402398 [ Links ]
4. Feben C, Kromberg J, Wainwright R, et al. Phenotypic consequences in black South African Fanconi anaemia patients homozygous for a founder mutation. Genet Med 2014;16(5):400-406. https://doi.org/10.1038/gim.2013.159 [ Links ]
5. Feben C, Kromberg J, Wainwright R, et al. Haematological consequences of a FANCG founder mutation in black South African patients with Fanconi anaemia. Blood Cells Mol Dis 2015;54(3):270-274. https://doi.org/10.1016/j.bcmd.2014.11.011 [ Links ]
6. Feben C, Haw T, Stones D, et al. Fanconi anaemia in South African patients with Afrikaner ancestry. S Afr J Child Health 2017;11(3):141-145. https://doi.org/10.7196/SAJCH.2017.v11i2.1312 [ Links ]
7. Shimamura A, Alter B. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 2010;24(10):101-133. https://doi.org/10.1016/j.blre.2010.03.002 [ Links ]
8. National Cancer Institute. Dictionary of genetic terms. https://www.cancer.gov/publications/dictionaries/genetics-dictionary (accessed 6 June 2017). [ Links ]
9. Giampietro PF, Adler-Brecher B, Verlander PC, et al The need for more accurate and timely diagnosis in Fanconi anaemia: A report from the International Fanconi Anaemia Registry. Pediatrics 1993;91(6):1116-1120. [ Links ]
10. Sawyer SL, Tian L, Kahkonen M, et al. Biallelic mutations in BRCA1 cause a new Fanconi anaemia subtype. Cancer Discov 2015;5(2):135-142. https://doi.org/10.1158/2159-8290.cd-14-1156 [ Links ]
11. Kim H, d'Andrea AD. Regulation of DNA cross-link repair by the Fanconi anaemia/BRCA pathway. Genes Dev 2012;26(13):1393-1408. https://doi.org/10.1101/gad.195248.112 [ Links ]
12. De Winter JP, Joenje H. The genetic and molecular basis of Fanconi anaemia. Mutat Res 2009;668(1-2):11-19. https://doi.org/10.1016/j.mrfmmm.2008.11.004 [ Links ]
13. Ameziane N, May P, Haitjema A, et al. A novel Fanconi anaemia subtype associated with a dominantnegative mutation in RAD51. Nat Commun 2015;6:8829. https://doi.org/10.1038/ncomms9829 [ Links ]
14. Bluteau D, Masliah-Planchon J, Clairmont C, et al. Biallelic inactivation of REV7 is associated with Fanconi anaemia. J Clin Invest 2016;126(9):2580-3584. https://doi.org/10.1172/JCI88010 [ Links ]
15. Park JY, Virts EL, Jankowska A, et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet 2016;53(10):672-680. https://doi.org/10.1136/jmedgenet-2016-103847 [ Links ]
16. Knies K, Inano S, Ramirez MJ, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anaemia. J Clin Invest 2017;127(8):3013-3027. https://doi.org/10.1172/JCI92069 [ Links ]
17. Tischkowitz M, Xia B. PALB2/FANCN - recombining cancer and Fanconi anaemia. Cancer Res 2010;70(19):7353-7359. https://doi.org/10.1158/0008-5472.CAN-10-1012 [ Links ]
18. Feben C, Spencer C, Lochan A, et al. Biallelic BRCA2 mutations in two black South African children with Fanconi anaemia. Fam Cancer 2017;16(3):441-446. https://doi.org/10.1007/s10689-017-9968-y [ Links ]
19. Demuth I, Wlodarski M, Tipping AJ, et al. Spectrum of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur J Hum Genet 2000;8(11):861-868. https://doi.org/10.1038/sj.ejhg.5200552 [ Links ]
20. Auerbach AD, Greenbaum J, Pujara K, et al. Spectrum of sequence variation in the FANCG gene: An International Fanconi Anaemia Registry (IFAR) study. Hum Mutat 2003;21(2):158-168. https://doi.org/10.1002/humu.10166 [ Links ]
21. Gillio AP, Verlander PC, Batish SD, et al. Phenotypic consequences of mutations in the Fanconi anaemia FAC gene: An International Fanconi Anaemia Registry Study. Blood 1997;90(1):105-110. [ Links ]
22. Wainstein T, Kerr R, Mitchell CL, et al. Fanconi anaemia in black South African patients heterozygous for the FANCG c.637 643delTACCGCC founder mutation. S Afr Med J 2013;103(12 Suppl 1):970-973. https://doi.org/10.7196/SAMJ.7215 [ Links ]
23. Whitney MA, Saito H, Jakobs PM, et al. A common mutation in the FACC gene causes Fanconi anaemia in Ashkenazi Jews. Nat Genet 1993;4(2):202-205. https://doi.org/10.1038/ng0693-202 [ Links ]
24. De Kerviler E, Guermazi A, Zagdanski E, et al. The clinical and radiological features of Fanconis anaemia. Clin Radiol 2000;55(5):340-345. https://doi.org/10.1053/crad.2000.0445 [ Links ]
25. Petryk A, Kanakatti Shankar R, Giri N, et al. Endocrine disorders in Fanconi anaemia: Recommendations for screening and management. J Clin Endocrinol Metab 2015;100(3):803-811. https://doi.org/10.1210/jc.2014-4357 [ Links ]
26. Kutler DI, Singh B, Satagopan J, et al. A 20-year perspective on the International Fanconi Anaemia Registry (IFAR). Blood 2013;101(4):1249-1256. https://doi.org/10.1182/blood-2002-07-2170 [ Links ]
27. Tischkowitz MD, Hodgson SV. Fanconi anaemia. J Med Genet 2003;40(1):1-10. https://doi.org/10.1136/jmg.40.1.1 [ Links ]
28. Auerbach AD. Fanconi anaemia and its diagnosis. Mutat Res 2009;668(1-2):4-10. https://doi.org/10.1016/j.mrfmmm.2009.01.03 [ Links ]
29. Alter BP. Fanconi's anaemia and malignancies. Am J Hematol 1996;53(2):99-1101. https://doi.org/10.1002/(SICI)1096-8652(199610)53:2<99::AID-AJH7>3.0.CO;2-Z [ Links ]
30. Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anaemia. Blood 2003;101(3):822-826. https://doi.org/10.1182/blood-2002-05-1498 [ Links ]
31. Hirsch B, Shimamura A, Moreau L, et al. Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood 2004;103(7):2554-2559. https://doi.org/10.1182/blood-2003-06-1970 [ Links ]
32. Wagner JE, Tolar J, Levran O, et al. Germline mutations in BRCA2: Shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anaemia. Blood 2004;103(8):3226-3229. https://doi.org/10.1182/blood-2003-09-3138 [ Links ]
33. Reid S, Renwick A, Seal S, et al., for the Familial Wilms Tumour Collaboration. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet 2005;42(2):147-151. https://doi.org/10.1136/jmg.2004.022673 [ Links ]
34. Meyer S, Tischkowitz M, Chandler K, et al. Fanconi anaemia, BRCA2 mutations and childhood cancer: A developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J Med Genet 2014;51(2):71-75. https://doi.org/10.1136/jmedgenet-2013-101642 [ Links ]
35. Antoniou AC, Casadei S, Heikkinen T, et al. Breast cancer risk in families with mutations in PALB2. N Engl J Med 2014;371(17):497-506. https://doi.org/10.1056/NEJMc1410673 [ Links ]
36. Sopik V, Akbari MR, Narod SA. Genetic testing for RAD51C mutations: In the clinic and community. Clin Genet 2015;88(4):303-312. https://doi.org/10.1111/cge.12548 [ Links ]
37. Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene 2006;25(43):5875-5884. https://doi.org/10.1038/sj.onc.1209878 [ Links ]
38. Neveling K, Endt D, Hoehn H, et al. Genotype-phenotype correlations in Fanconi anaemia. Mutat Res 2009;668(1-2):73-91. https://doi.org/10.1016/j.mrfmmm.2009.05.006 [ Links ]
39. Nicchia E, Greco C, de Rocco D, et al. Identification of point mutations and large intragenic deletions in Fanconi anaemia using next generation sequencing technology. Mol Genet Genomic Med 2015;3(6):500-512. https://doi.org/10.1002/mgg3.160 [ Links ]
40. Christianson A, Howson C, Modell B. March of Dimes Global Report on Birth Defects: The hidden toll of dying and disabled children. 2006. https://www.marchofdimes.org/materials/global-report-on-birth-defects-the-hidden-toll-of-dying-and-disabled-children-full-report.pdf (accessed 16 October 2017). [ Links ]
41. MacMillan ML, Wagner JE. Haematopoietic stem cell transplantation for Fanconi anaemia - when and how? Br J Haematol 2010;149(1):4-21. https://doi.org/10.1111/j.1365-2141.2010.08078.x [ Links ]
42. Tischkowitz M, Dokal I. Fanconi anaemia and leukaemia - clinical and molecular aspects. Br J Haematol 2004;126(2):176-191. https://doi.org/10.1111/j.1365-2141.2004.05023.x [ Links ]
43. Dufour C, Svahn J. Fanconi anaemia: New strategies. Bone marrow transplant 2008;41:S90-S95. https://doi.org/10.1038/bmt.2008.63 [ Links ]
44. Frohnmayer D, Frohnmayer L, Guinan E, Kennedy T, Larsen K, eds. Fanconi Anaemia: Guidelines for Diagnosis and Management. 4th ed. Fanconi Anemia Research Fund, 2014. https://fanconi.org/images/uploads/other/Guidelines_4th_Edition.pdf (accessed 15 April 2018). [ Links ]
45. Zierhut HA, Tryon R, Sanborn EM. Genetic counseling for Fanconi anemia: Crosslinking disciplines. J Genet Couns 2014;23(6):910-921. https://doi.org/10.1007/s10897-014-9754-z [ Links ]
46. Berliner JL, Fay AM, Cummings SA, et al. NSGC Practice Guideline: Risk assessment and genetic counseling for hereditary breast and ovarian cancer. J Genet Couns 2013;22(2):155-163. https://doi.org/10.1007/s10897-012-9547-1 [ Links ]
47. Zierhut HA, Bartels DM. Waiting for the next shoe to drop: The experience of parents of children with Fanconi anemia. J Genet Couns 2012;21(1):45-58. https://doi.org/10.1007/s10897-011-9394-5 [ Links ]
48. Faivre L, Guardiola P, Lewis C, et al. Association of complementation group and mutation type with clinical outcome in Fanconi anaemia. Blood 2000;96(13):4064-4070. [ Links ]
 Correspondence:
Correspondence:
C Feben
candice.feben@nhls.ac.za
Accepted 8 January 2018


{kind=link}
{kind=link}
{kind=link}