








Serviços Personalizados
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkSAMJ: South African Medical Journal
versão On-line ISSN 2078-5135
versão impressa ISSN 0256-9574
SAMJ, S. Afr. med. j. vol.111 no.6 Pretoria Jun. 2021
http://dx.doi.org/10.7196/SAMJ.2021.v111i6.15724
CME
N A AliiI; M PatelII; J PoolIII; Y GogaIV; A KrauseV
IMB BCh, FCPath (SA) Haem; Department of Molecular Medicine and Haematology, Faculty of Health Sciences, University of the Witwatersrand and National Health Laboratory Service, Johannesburg, South Africa
IIMB ChB, FCP (SA), MMed (Haem), FRCP (Lond), PhD; Clinical Haematology Unit, Department of Medicine, Chris Hani Baraggwanath Academic Hospital and Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
IIIMB BCh, DCH (SA), FCPaed (SA); Department of Paediatrics and Child Health, Faculty of Health Sciences, University of the Witwatersrand and Charlotte Maxeke Academic Hospital, Johannesburg, South Africa
IVMB BCh, DCH (SA), FCPaed (SA), Cert Clin Haematology (SA) Paed, MSc (Bioethics), PG Dip Paed Pall Medicine; Inkosi Albert Luthuli Central Hospital, and Department of Paediatrics and Child Health, Nelson R Mandela School of Medicine, University of KwaZulu-Natal, Durban, South Africa
VMB BCh, PhD; Division of Human Genetics, National Health Laboratory Service and School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Johannesburg, South Africa
ABSTRACT
The thalassaemias are a group of inherited blood disorders characterised by decreased or absent globin chain synthesis. Classification of thalassaemia is based on the type of globin chain that is deficient. There are four globin chain subtypes, viz. α, β, γ and δ; deficiencies of these are designated as α-, β-, γ- or δ-thalassaemia, respectively. As genetic defects or deletions may affect one or more globin genes, a variety of clinical phenotypes may be encountered. Heterozygotes (thalassaemia carriers) enjoy partial protection against malaria, with consequential survival advantage. This explains the unexpectedly high gene frequencies found in malaria-endemic areas. South Africa is not a malaria-endemic area, and therefore has a predictably low prevalence of thalassaemia and other inherited Hb disorders. However, because of migration, these conditions are increasingly encountered in countries not endemic to malaria. In this issue (part 1 of the 2-part CME series), discussion is centred around epidemiology, pathophysiology, clinical features and diagnosis. Management is discussed in the next issue (part 2).
This CME series on thalassaemia is divided into 2 sections: part 1 (current issue) covers epidemiology, pathophysiology, clinical exposition and diagnosis, whereas part 2 (next issue) is dedicated to matters pertaining to management.
The term thalassaemia is derived from the Greek word thalassa, which means sea, as thalassaemia was known for its prevalence around the Mediterranean Sea.
The thalassaemias are a group of inherited disorders that result in a reduction or absence of globin chain synthesis. Classification of thalassaemia is based on the type of globin chain that is deficient or absent, viz. α-, β-, γ-, δ-, δβ-, γδβ- and εγδβ-thalassaemia.
Human Hb is a tetrameric molecule that is made up of two α- and two non-α-globin chains. For Hb to function optimally, there must be equal representation of α- and non-α-globin chains. Postnatally there are 3 Hb subtypes, i.e. HbA, HbA2 and HbF (Table 1).

Since HbA is the major Hb subtype that constitutes >95% of total Hb after infancy, α- and β-thalassaemias represent the majority of clinically important thalassaemias. Similarly, with HbF being the major subfraction in the fetus, γ-thalassaemia is likely to cause varying degrees of anaemia in the fetus, depending on the number of γ-globin genes inactivated or deleted. In contrast, as the α-globin chain is an integral component of all Hb subtypes after the embryonic phase, α-thalassaemia manifests in the fetus, as well as postnatally (children and adults).
Alpha-globin chains are transcribed on chromosome 16 (Fig. 1) and non-α-globin ' chains are transcribed on chromosome 11 (Fig. 2) (discussed below). Delta-beta-thalassaemia is caused by deletion of a δ-gene and the adjacent β-globin gene. Larger deletions may include ther globin genes and cause γδβ- and εγδβ-thalassaemia, which are uch rarer conditions (Fig. 2). Gamma- and δ-thalassaemia do not ause any clinically recognisable disease postnatally, as HbA2 and bF constitute only a small fraction of Hb in adults.


Owing to the rarity of δβ-, γδβ- and εγδβ-thalassaemia, and the linical insignificance of γ- and δ-thalassaemia, these entities are not discussed further in this CME series.
Epidemiology and genetic basis of disease
Most inherited red cell disorders, including thalassaemia, owe their high prevalence in malaria-endemic regions to selective pressure of malaria. Partial protection of heterozygotes (thalassaemia carriers) against malaria with consequent natural selection has been responsible for elevating and maintaining their gene frequencies, an idea first proffered 50 years ago by Haldane[1,2] Several mechanisms for reduced parasite survival have been proposed and the oxidative stress pathway has been reported to be a likely mechanism of protection. Oxidative stress of the parasite is added to the intrinsic oxidative stress of the thalassaemic red cell, thereby decreasing its overall viability. This allows for selective removal of the parasitised red cell by macrophages.[3-5] However, individuals with severe forms of thalassaemia are particularly sensitive to malarial infection.
The mode of inheritance of severe forms of thalassaemia is typically autosomal recessive.
Alpha-thalassaemia
Alpha-thalassaemia is subdivided into three main categories:
• Inherited α-thalassaemia syndromes (Table 2). These constitute the most common variety and are caused by deletions (usually) or mutations in and around the α-globin gene cluster that silence or remove one or more α-globin genes.
• Alpha-thalassaemia mental retardation syndrome, a rare condition of which there are two distinct genetic subtypes:[6]
• ATR-X syndrome, caused by a mutation in the ATRX gene on the X-chromo-some (Xq13.1 - q21.1). This syndrome manifests with severe mental retardation, congenital organ and facial anomalies and mild thalassaemia. It is inherited in an X-linked recessive manner.
• ATR-16 syndrome, caused by a micro-deletion of the tip of chromosome 16p. This disorder is associated with a milder level of intellectual disability and more subtle facial and genital anomalies. Depending on the size of the deletion, patients may also share features of other conditions, including polycystic kidneys and tuberous sclerosis.
In both subtypes, the α-thalassaemia component does not cause clinically significant haemolysis, but detection of a low number of red cell HbH inclusions, or mild microcytic hypochromic indices generally serve as a supportive diagnostic tool for the identification of such cases.
• Alpha-thalassaemia myelodysplastic syndrome (ATMDS) is very rare, and has been described in select haemato-logical malignancies, including myelodys-plastic syndrome (MDS), myeloproliferative disorders and erythroleukaemia.[7-11] Such patients usually present with severe anaemia as a result of uncompensated haemolysis and generally require regular transfusions.
Discussion of α-thalassaemia is focused on the common inherited variety, which is not associated with any syndromic features.
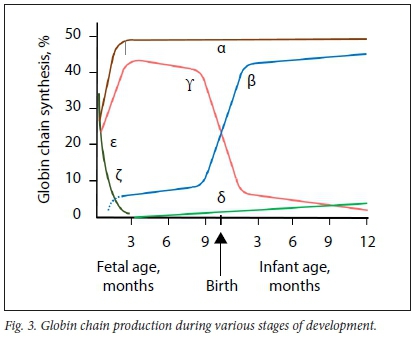
The α-globin cluster is situated on the short arm of chromosome 16 (16pl3.3) and α-globin chains are encoded by two closely linked genes on each chromosome (Fig. 1). Thus, four genes encode the α-globin chain, two on each chromosome 16. HS40 is a regulatory gene that is responsible for co-ordinated and sequential expression of the entire α-globin cluster during development. The ζ-globin gene is an α-like gene that is expressed during the embryonic phase until ~8 weeks of fetal development, followed by a switch in expression to α-globin genes. Expression of α-globin genes continues throughout adult life (Figs 3 and 4).
The normal genotype is designated as αα/ αα, indicating the presence of four α-genes. Alpha-thalassaemia is classified as α0, with deletion or mutation of both α-genes on a linked pair (- -/αα), or α+-thalassaemia, with deletion or mutation of one α-gene on a linked pair (-α/αα). If three of the α-genes are deleted, the resultant disorder is called HbH disease. Deletion of all four α-genes is a lethal abnormality, and causes severe fetal anaemia and hydrops fetalis.
Alpha-thalassaemia is widely distributed geographically, largely reflecting areas of malaria endemicity, ranging from sub-Saharan Africa to the Mediterranean basin, Arabian Peninsula, Indian subcontinent and South East Asia in a line crossing through southern China, Thailand, Malaysia, Indonesia and the Pacific islands. The α+-mutation (discussed below) is the predominant type in Africa, where the frequency ranges from 0.12 to 0.4 across various parts of the continent.[12-15] A more recent study, investigating the role of α-thalassaemia in unexplained microcytosis in the South African (SA) population, demonstrated the prevalence of α-thalassaemia trait to be 19.1% among black and Indian patients.[16] The -α3.7 deletion is the most common deletion identified in SA. Other deletions reflect the ancestral origins of individuals.[16,17]
Beta-thalassaemia
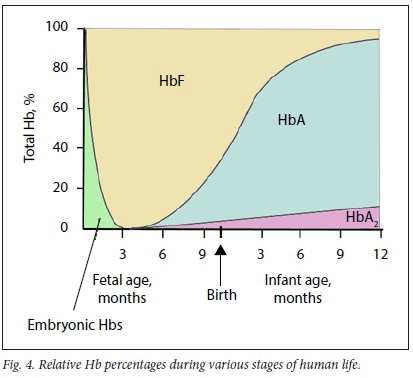
The β-globin cluster is situated on the short arm of chromosome 11 (1 lpl5.5). On each chromosome, £-, δ- and β-globin chains are each encoded by a single gene, whereas γ-globin chains are encoded by two genes. Therefore, in a normal person, two gene copies of each encode the ε-, δ- and β-chains, whereas four gene copies encode the γ-globin chains (Fig. 2). The locus control region (LCR) controls the expression of the non-a-globin genes sequentially during development in the order in which they appear on the chromosome. The ε-globin gene is expressed during the embryonic phase until ~8 weeks of fetal development. Thereafter, expression is switched to γ-globin genes until -30 weeks of fetal lite, when another switch to β-globin gene expression occurs. Low-level production of γ-globin chains, however, continues throughout adult life. Transcription of δ-globin genes is inefficient, which explains the low level of HbA2 (2.4 - 3.7%) (Fig. 3). From 6 to 12 months of life onwards, HbA is the predominant Hb subtype, whereas HbF and HbA2 (as minor haemoglobins) constitute <1% and 2.4 - 3.7% of total Hb, respectively (Fig. 4).
The most common mutations of the β-globin gene are point mutations and small insertions or deletions of one or two bases. Deletions of the β-globin gene are generally uncommon, although a 619bp deletion occurs at a frequency of up to 12% in SA individuals of Indian ancestry with thalassaemia.1171 Individuals with mutations in both β-globin genes have β-thalassaemia major, and suffer from severe and often uncompensated haemolytic anaemia. Individuals with one mutant gene copy are so-called carriers. They are generally asymptomatic, but have microcytic hypochromic red cell indices.
The geographical distribution shows significant overlap with that of a-thalassaemia and includes the Mediterranean region, segments of North and West Africa, the Middle East, Indian subcontinent and South East Asia.
• Asymptomatic thalassaemia trait: (i) heterozygous ß-thalassaemia; and (ii) single-gene (-α/αα) or two-gene deletion ((- -/αα) or (-α/-α)) α-thalassaemia. Although asymptomatic, it is important to identify such individuals, as they are often misdiagnosed as having iron-deficiency anaemia and unnecessarily supplemented with iron.
• Thalassaemia intermedia: in this category there is chronic haemolysis that is compensated, i.e. not necessitating regular transfusions. The Hb is maintained at a steady level with moderate anaemia. Such patients may be diagnosed only when they develop aplastic crisis with severe pallor, or hyperhaemolysis with jaundice due to intercurrent infection. However, as a result of the chronic anaemia and hypoxia, long-term effects of chronic haemolysis are likely to develop. Examples in this category include HbH disease (-/-α) and homozygous ß+-thalassaemia (discussed below).
• Thalassaemia major: such patients have severe haemolysis that is not compensated by the bone marrow and are thus transfusion dependent. They require aggressive management and close monitoring to prevent the complications of chronic haemolysis and hypoxia. Presenting features of thalassaemia major include severe anaemia, jaundice, dark urine, failure to thrive, stunted growth and effects of chronic haemolysis, such as splenomegaly, and bony abnormalities (Table 3). A characteristic example of thalassaemia major is homozygous ß0-thalassaemia (discussed below).
The long-term effects and complications of chronic haemolytic anaemia are listed in Table 3. With adequate treatment and compliance, the complication rate and severity can be prevented or minimised. Management aspects are discussed in part 2 of this CME series.
Alpha-thalassaemia
The clinical presentation is variable and depends on the number of α-globin genes that are deleted or inactivated.
Patients with deletions of one or two α-globin genes are generally asymptomatic and have no long-term clinical sequelae. The mean cell volume (MCV) and mean cell Hb (MCH) are decreased, the Hb is usually within or at the lower limit of the normal range and the red cell count (RCC) is normal or elevated.
Deletion of three α-globin genes (- -/-α)), also known as HbH disease, falls under the category of thalassaemia intermedia. Reduced α-chain production results in a relative excess of γ-chains (during fetal life) and ß-chains (postnatally). Excess γ- and ß-chains have the ability to form γ4- and ß4-tetramers, named Hb Barts and HbH, respectively. Hb Barts and HbH are unstable, and consequently cause mature red cells to haemolyse. Fortunately, owing to the capacity of the bone marrow to increase its output 6-fold, a steady-state Hb level of ~8 - 9 g/dL is usually maintained (compensated haemolysis). However, a minority of patients with HbH disease require long-term transfusions, as they are unable to maintain a steady-state Hb (uncompensated haemolysis). In addition, Hb Barts and HbH both have very high oxygen affinities and are of little use for oxygen delivery to tissues.
Deletion of all four α-globin genes (Hb Barts hydrops fetalis) results in complete absence of α-chain production, a scenario that is incompatible with life since the α-chain is an integral component of HbF and HbA. Fetuses become hydropic owing to severe in utero anaemia, with resultant cardiac failure, and generally death in the second or third trimester of pregnancy.
Beta-thalassaemia
Depending on the mutation, the output of the affected ß-globin gene may be decreased (designated as ß+) or completely absent (designated as ß0). Heterozygous ß-thalassaemia, whether ß+ or ß0, is generally asymptomatic, with no adverse clinical consequences. The MCV is decreased and the Hb is usually within the normal or lower limit of the normal range. The RCC is typically at the upper limit of the normal range or increased.
Homozygous ß0-thalassaemia presents with severe anaemia, which is transfusion dependent. Infants are asymptomatic at birth due to high enough levels of HbF. As the HbF level falls during the first few months of life, there is no reciprocal increase in HbA production, causing severe anaemia 3 - 6 months after birth.
Deficiency or absence of ß-globin chains results in a relative excess of α-globin chains which, unlike β- and Y-chains, cannot form tetramers. α-chain monomers are insoluble and precipitate in the red cell, causing membrane damage and haemolysis prior to release from the bone marrow, referred to as 'ineffective erythropoiesis'.
The presenting clinical features include pallor with progressive anaemia, poor growth, difficulty in feeding, cardiac failure, hepatosplenomegaly and recurrent infections.[18] In patients who are inadequately transfused, the bone marrow cavity expands in an attempt to increase haemopoietic capacity, which results in skeletal abnormalities (including maxillary hyperplasia and frontal bossing).
In homozygous ß+-thalassaemia, ß-globin chains are produced with 10 - 30% efficiency. Haemolysis occurs to a moderate degree and is compensated. The disease runs a milder clinical course, with no transfusion dependency, and falls into the category of thalassaemia intermedia. These patients are nonetheless susceptible to the long-term effects of chronic haemolysis and anaemia (Table 3).
Diagnosis
Thalassaemia is often suspected on the full blood count (FBC) with blood-smear microscopy, clinical examination or a known family history. In most instances, the diagnosis is obtained using conventional Hb separation techniques, such as high-performance liquid chromatography (HPLC), Hb electrophoresis or capillary electrophoresis (Table 4).
Heterozygous α- or ß-thalassaemia is usually detected incidentally on FBC testing or in the course of family studies. An important indication is microcytosis and hypochromia in the absence of iron deficiency and anaemia of chronic disorder. The MCV/RCC ratio, known as the Mentzer index, is a useful tool to distinguish iron deficiency and anaemia of chronic disorder from heterozygous thalassaemia, where a ratio of <13 is indicative of the latter. The Mentzer index has a sensitivity and specificity of 98.7 and 82.3%, respectively.[19,20] In addition, the red cell distribution width is generally normal in thalassaemia trait as opposed to iron deficiency, where it is usually raised. Whereas the HbA2 level is normal to decreased in α-thalassaemia trait, it is typically raised in ß-thalassaemia trait and is a diagnostic hallmark of the condition (Table 4). In ß-thalassaemia major and HbH disease, peripheral blood microscopy shows marked red cell changes.
Incubation of red cells in a reticulocyte preparation for 2 - 3 hours causes HbH (ß4) to precipitate and form lattice-like inclusions within the red cells. These are called 'golf balls' due to their appearance, and are easily detected under light microscopy. Scanty HbH inclusions may be observed in α-thalassaemia trait, but are found in abundance in HbH disease.[21]
Genetic analysis for α- and ß-thalassaemia mutations requires somewhat different approaches due to the different types of causative mutations. For α-thalassaemia, initial testing uses a multiplex polymerase chain reaction (PCR) analysis that detects the most common deletions. This confirms the diagnosis in most instances.[22]
The majority of β-thalassaemia mutations are point mutations or small insertions/ deletions. Sequencing of the complete β-globin gene detects most mutations. It is important to ensure that introns are also sequenced, as several relatively common mutations are situated deep within the introns. Gene sequencing has typically been done by Sanger sequencing, but next-generation sequencing (NGS) is increasingly being used. Analysis for large deletions may be required, particularly if a δβ-thalassaemia is suspected.
Genetic diagnosis can be used where there is diagnostic uncertainty after haematological analysis. Further, prenatal or pre-implantation diagnosis can be offered to couples who are at high risk of having a child with a severe haemoglobinopathy.[23]
As the majority of severe haemoglobinopathies are inherited in autosomal recessive fashion, there is a risk of 25% (1/4) - if both parents are carriers - to have a child with a severe haemoglobinopathy. The risk is the same with every pregnancy. Parents may therefore choose prenatal diagnosis during a pregnancy. This requires analysis of fetal DNA obtained by amniocentesis (performed from 16 to 20 weeks of gestation) or chorionic villus sampling (performed from 11 to 13 weeks of gestation). Ideally, the mutations in the parents should be determined prior to the pregnancy, so that testing can be performed rapidly once fetal DNA is obtained.
Pre-implantation genetic testing (PGT) can also be performed for haemoglobinopathies, which requires the testing of trophoblastic cells obtained from embryos produced by in vitro fertilisation (IVF). Parental mutations need to be identified in advance of the prenatal testing procedure. PGT is performed in laboratories specifically set up to test embryos and not in routine genetic testing laboratories. After testing, unaffected embryos may be returned to the uterus in the hope of achieving a successful pregnancy. Pre-implantation genetic diagnosis saves parents from having to make a decision regarding termination of an affected pregnancy. PGT is, however, a costly process, as it requires IVF and specialised testing. A number of IVF cycles may be required before a pregnancy is achieved.[24] Couples may also choose to test embryos simultaneously for human leukocyte antigen (HLA) compatibility, where an affected child could be assisted by a stem cell transplant from an unaffected sibling. However, selection of unaffected and HLA-matched embryos greatly reduces the number of embryos suitable for implantation.
Declaration. None.
Acknowledgements. None.
Author contributions. NAA: design and content; MP: content; JP: content; YG: content; and AK: content.
Funding. None.
Conflicts of interest. None.
References
1. Hendrick PW. Population genetics of malaria resistance in humans. Heredity 2011;107(4):283-304. https://doi.org/10.1038/hdy.2011.16 [ Links ]
2. Haldane JBS. The rate of mutation of human genes. Hereditas 1949b;35(Suppl 1):267-273. [ Links ]
3. Attama H, Ginsburg H. The malaria parasite supplies glutathione to its host cell - investigation of glutathione transport and metabolism in human erythrocytes infected with Plasmodium falciparum. Eur J Biochem 1997;250(3) :670-679. https://doi.org/10.1111/j.1432-1033.1997.00670.x [ Links ]
4. Rachmilewitz EA, Shinar E, Shalev O, Galili U, Schrier SL. Erythrocyte membrane alterations in beta-thalassaemia. Clin Hematol 1985;14(1):163-182. [ Links ]
5. Schrier SL. Pathobiology of thalassemic erythrocytes. Curr Opin Hematol 1997;4(2):75-78. https://doi.org/10.1097/00062752-199704020-00001 [ Links ]
6. Gibbons RJ, Higgs DR. The α-thalassaemia/mental retardation syndromes. Medicine (Baltimore) 1996;75:45-52. [ Links ]
7. Boehme WM, Piira TA, Kurnick JE, Bethlerfalvay NC. Acquired hemoglobin H in refractory sideroblastic anemia. A preleukaemic marker. Arch Intern Med 1978;138:603. [ Links ]
8. Yoo D, Schechter GP, Amigable AN, Nienhuis AW. Myeloproliferative syndrome with sideroblastic anemia and acquired hemoglobin H disease. Cancer 1980;45( 1):78-83. https://doi.org/10.1002/1097-0142(19800101)45:1<78::aid-cncr2820450114>3.0.co;2-n [ Links ]
9. Agarwal MB, Agarwal UM, Pai BM. Acquired haemoglobin H disease in a case of myelodysplastic syndrome. J Assoc Physicians India 1991;30(10):769-770. [ Links ]
10. Abbondanzo SL, Anagnou NP, Sacher RA. Myelodysplastic syndrome with acquired hemoglobin H disease. Evolution through megakaryoblastic transformation into myelofibrosis. Am J Clin Pathol 1988;89(3):401-406. [ Links ]
11. Annino L, di Giovanni S, Tentori L Jr, et al. Acquired hemoglobin H disease in a case of refractory anemia with excess blasts (RAEB) evolving into acute nonlymphoid leukaemia. Acta Haematol 1994;72(1):41-44. [ Links ]
12. Dodé C, Berth A, Rochette J, Girot R, Labie D. Analysis of crossover type in the alpha'3,7 haplotype among sickle cell anemia patients from various parts of Africa. Hum Genet 1988;78(2):193-195. https://doi.org/10.1007/BF00278197 [ Links ]
13. Ramsay M, Jenkins T. Alpha-thalassaemia in Africa: The oldest malaria protective trait? Lancet 1984;2(8399):410. https://doi.org/10.1016/s0140-6736(84)90581-6 [ Links ]
14. Bird AR, Ellis P, Wood K, Mathew C, Karabus C. Inherited haemoglobin variants in a South African population. J Med Genet 1987;24(4):215-219. https://doi.org/10.1136/jmg.24A215 [ Links ]
15. Mears JG, Lachman HM, Labie D, Nagel RL. Alpha-thalassemia is related to prolonged survival in sickle cell anemia. Blood 1983;62(2):286-290. [ Links ]
16. Loonat SB, Naran NH, Thein SL, Alli NA. Alpha-thalassaemia trait as a cause of unexplained microcytosis in a South African population. S Afr Med J 2016;106(3):276-279. https://doi.org/10.7196/SAMJ.2016.v106i3.10005 [ Links ]
17. Krause A, Wainstein T, Essop FB, Goodyear Q. Testing for haemoglobinopathies in Johannesburg, South Africa: A 30-year review. S Afr Med J 2013;103(12 Suppl 1):989-993. https://doi.org/10.7196/samj.7255 [ Links ]
18. Nithichanon A, "Tissakhon I, Samer W, et al Immune responses in beta-thalassaemia: Heme oxygenase 1 reduces cytokine production and bactericidal activity of human leucocytes. Sci Rep 2020;10(1):10297. https://doi.org/10.1038/s41598-020-67346-2 [ Links ]
19. Vehapoglu A, Ozgurhan G, Demir AD, et al. Hematological indices for differential diagnosis of beta thalassemia trait and iron deficiency anemia. Anemia 2014:576738. https://doi.org/10.1155/2014/576738 [ Links ]
20. Herbert L, Muncie JR, Campbell JS. Alpha and beta thalassaemia. Am Fam Phys 2009;80(4):339-344. [ Links ]
21. Chan LC, So JCC, Chui DHK. Comparison of haemoglobin H inclusion bodies with embryonic ζ globin in screening for α thalassaemia. J Clin Pathol 1995;48:861-864. https://doi.org/10.1136/jcp.48.9.861 [ Links ]
22. Chong SS, Boehm CD, Higgs DR, Cutting GR. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood 2000;95(1):360-362. https://doi.org/10.1182/blood.V95.L360 [ Links ]
23. Origa R. Beta-thalassemia. GeneReviews 2000. https://www.ncbi.nlm.nih.gov/books/NBK1426/ (accessed 29 April 2021). [ Links ]
24. Carzis B, Wainstein T, Gobetz L, Krause A. Review of 10 years of preimplantation genetic diagnosis in South Africa: Implications for a low-to-middle-income country. J Assist Reprod Genet 2019;36(9):1909-1916. https://doi.org/10.1007/s10815-019-01537-3 [ Links ]
 Correspondence:
Correspondence:
N A Alli
nazeer.alli@nhls.ac.za
Accepted 29 March 2021

{kind=link}
{kind=link}
{kind=link}