










Services on Demand
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
Indicators

Related links
-
 Cited by Google
Cited by Google -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkSouth African Journal of Chemistry
On-line version ISSN 1996-840X
Print version ISSN 0379-4350
S.Afr.j.chem. (Online) vol.66 Durban Aug. 2013
RESEARCH ARTICLE
An electrochemical investigation of methanol oxidation on nickel hydroxide nanoparticles
Jahan Bakhsh Raoof*; Reza Ojani; Sayed Reza Hosseini
Department of Analytical Chemistry, Faculty of Chemistry, University of Mazandaran, 3rd Kilometer of Air Force Road, 47416-95447, Babolsar, Iran
ABSTRACT
In this work, a nickel-modified glassy carbon electrode (GCE) was prepared using a potentiostatic method in 0.1 M acetate buffer solution at pH 4.0 containing 1.0 mM nickel nitrate. Nickel hydroxide nanoparticles were prepared using consecutive potential scanning in 0.1 M NaOH solution. The as-prepared catalyst was characterized by field emission scanning electron microscopy and electrochemical methods. Electrochemical characterization exhibited stable redox behaviour of the Ni(III)/Ni(II) couple. Cyclic voltammetric experiments showed that electrocatalytic oxidation of methanol can occur at the modified electrode, whereas it is not observed at a bare GCE. The effect of potential sweep rates and methanol concentration on its electrochemical behaviour was studied. The rate constant (k) for the chemical reaction between methanol and catalytic centres has been evaluated by chronoamperometry. In addition, long-term stability of the modified electrode was investigated by electrochemical methods.
Keywords: Cyclic voltammetry, electrooxidation, glassy carbon electrode, methanol, nickel hydroxide nanoparticles.
1. Introduction
The electrocatalytic oxidation of methanol is a topic of intensive research in the context of the development of direct methanol fuel cells as power sources for electric vehicles and electronic devices.1 In recent years, considerable efforts have been directed towards the study of methanol electrooxidation in solutions with high pH.2-4 The use of alkaline solutions in a fuel cell has many advantages such as increasing its efficiency,2,5,6 a wider selection of possible electrode materials, a better efficiency of oxygen cathode, and oxidation reactions of organic fuels exhibit almost no sensitivity to the surface structure.7 Smaller or negligible poisoning effects in alkaline solutions are observed.8 In this context, electrode materials are clearly important parameters in the electrochemical oxidation of methanol, where high efficient electrocatalysts are needed. Nickel as a low cost, relatively abundant material, has demonstrated long-term stability in alkaline solutions;9,10 hence, it is a useful catalyst for methanol oxidation.
Nickel oxides have been widely investigated due to their potential applications in electrochromic films, optical materials, fuel cell electrodes, photocatalysts, electrochemical capacitors, batteries, etc. Most of these useful functions depend mainly on the composition, the morphology and structure of the oxide deposits. Nanostructured nickel oxides are believed to have better properties than those of their bulk counterparts.11 There are many different methods reported for synthesis of the nanostructured nickel oxides.11-15 Some of them for preparing nickel oxide powder suffer from difficulty in fabrication of well-dispersed nickel oxide electrodes for electrochemical applications. Thus, it is more advantageous to have a porous electrode of nano-structured nickel oxide fabricated by electrochemical deposition, which deposits the active material directly onto the substrate at room temperature without templates.
Recently, we have combined the advantageous features of polymer modification and dispersion of nickel onto conducting polymers coated carbon paste electrodes by construction of Ni/P(1,5-DAN)/MCPE,16 Ni/P(OAP-SDS)/MCPE,17 Ni/P(OAP)/ MCPE,18 Ni/P(OT-TX-100)/MCNTPE19 and Ní/P(MT-CTAb)/ MCPE,20 which can successfully catalyze the methanol oxidation in alkaline medium. Our literature survey indicates that, there is no report as yet on the usage of nickel hydroxide nano-particles modified glassy carbon electrode (Ni(OH)2NPs/MGCE) for methanol oxidation. Thus, in this paper, with respect to advantages of nickel hydroxide nanoparticles and alkaline medium, we have decided to investigate the electrocatalysis and parameters affecting the electrocatalytic oxidation of methanol. The results show that the nanoparticles can electrooxidize the methanol with high current densities.
2. Experimental
2.1. Apparatus and Materials
Electrochemical experiments were performed with a computer controlled potentiostat/galvanostat µ-Autolab type III modular electrochemical system (Eco Chemie BV Netherlands), driven with general purpose electrochemical system (GPES) software (Eco Chemie). A conventional three electrode cell was used with Ag|AgCl|KCl (3 M) as reference electrode, platinum wire as counter electrode and glassy carbon disk (1.5 mm in diameter, Azar electrode Co., Iran) as working electrode substrate. The surface morphology of the deposit was evaluated by field emission scanning electron microscopy (FE-SEM) Hitachi S-4160. All experiments were carried out at ambient temperature.
Sodium hydroxide, sodium acetate, acetic acid and methanol used in this work were analytical grade of Merck origin and used without further purification. Ni(NO3)2∙6H2O and other reagents were analytical grade. All solutions were prepared with distillated water.
2.2. Fabrication of Ni(OH)2NPs/MGCE
Modification of GCE was performed according to procedure proposed by Salimi et al.21 Briefly, the GCE was polished with alumina slurries on a polishing cloth. Then, the electrode was placed in ethanol and sonicated to remove adsorbed particles. Nickel was initially electrodeposited (-1.0 V, 5.0 min deposition time) on the GCE from a 1.0 mM nickel nitrate in 0.1 M acetate buffer solution (pH 4.0). After this, the electrode was removed from the cell solution, washed with distilled water and then, cycled between 0.0 and 0.8 V for 200 cycles in 0.1 M NaOH solution. In the first anodic cycle, a small anodic peak appears which corresponds to the oxidation of metallic nickel particles according to Equation 1:22

This peak almost disappeared in the subsequent cycles.23 FE-SEM images show that the GCE is homogenously covered with Ni(OH)2 nanoparticles with average particle diameters less than 100 nm (Fig. 1).

3. Results and Discussion
3.1. Cyclic Voltammetry Behaviour
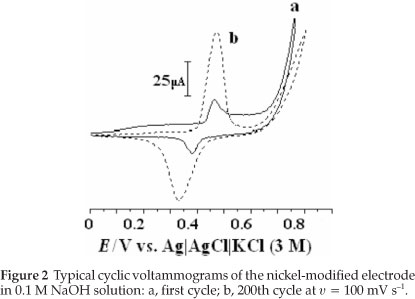
The polarization behaviour of the nickel-modified GCE was examined in 0.1 M NaOH solution by using cyclic voltammetry. Typical first and 200th cyclic voltammograms are represented in Fig. 2. Consecutive potential cycling leads to a progressive increase of anodic and cathodic peaks currents values. Changes in position of the peaks are likely due to changes in crystal structures of the nickel hydroxide and nickel oxyhydroxide constituents.24 Electrochemical behaviour corresponding to the anodic and cathodic peaks is attributed to the redox couple according to Equation 2:

3.2. Electrochemical Behaviour of the Ni(OH)2NPs/MGCE
Figure 3A shows the cyclic voltammograms obtained for the Ni(OH)2NPs/MGCE in 0.1M NaOH solution at different potential scan rates over a wide range, 0.005-1.0 V s-1. The anodic and cathodic peak currents are proportional to the potential scan rate (υ) at low values from 0.005 to 0.080 V s-1 (Fig. 3B). This can be attributed to an electrochemical activity of an immobilized redox couple at surface. From the slope of this line and using: 25


where Ip, A and I* are peak current, electrode surface area and surface coverage of the redox species, respectively, and taking the average of both cathodic and anodic currents, the total surface coverage of the immobilized active substance of about 3.6 x 10-8 mol cm-2 was derived.
3.3. Electrocatalytic Oxidation of Methanol at the
Ni(OH)2NPs/MGCE
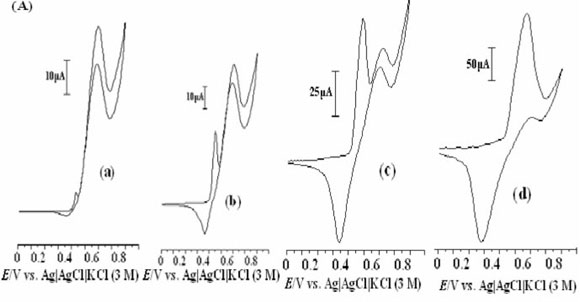
In this work, methanol oxidation was first studied at a bare GCE in 0.1 M NaOH solution (Fig. 4). The electrochemical response of the bare GCE in the absence of methanol is shown in curve (a); the addition of 0.04 M methanol to the alkaline solution causes no significant effect on the electrochemical response (curve (b)). Methanol oxidation at bare GCE in basic media is poor and it is not possible to obtain an oxidation peak prior to the discharge of the supporting electrolyte.

The electrochemical response of the Ni(OH)2NPs/MGCE exhibits well-defined anodic and cathodic peaks associated with the Ni(III)/Ni(II) redox couple (curve (c)). Curve (d) shows the behaviour of the modified electrode in the presence of 0.04 M methanol. An increment in the anodic peak current (aI) followed by appearance of a new peak (aII) at more positive potential and a decrease in the cathodic peak current and its charge (RI) during the reverse scan are main effects upon methanol addition to the electrolyte solution. The modifier layer Ni(OH)2 at the electrode surface acts as an electrocatalyst for methanol oxidation. It can be seen that the oxidation peak appears in two potentials regions. The electroreactive NiOOH species is generated from oxidation of Ni(OH)2 in the first region and reacts with methanol via chemical reaction and Ni(OH)2 is regenerated. Thus, there is an increase in a, peak current following a slow decrease in cathodic peak current in the negative scan of potential, i.e. an EC' mechanism:25,26

The relative decrease of the cathodic peak height in the presence of methanol is attributed to the partial consumption of NiOOH species in chemical reaction with methanol and Ni(OH)2 formation in accordance with Equation 5. This indicates clearly that the applied modifier in this process participates directly in electro-catalytic oxidation of methanol. On the basis of these observations, the catalytic role of Ni(III) for methanol oxidation is obvious as proposed previously by Fleischmann et al.27 In order to further clarify the oxidation mechanism onto the modified electrode, the effects of methanol concentration and potential scan rates on its voltammetric responses are investigated.
3.4. The Effect of Methanol Concentration
Figure 5 shows behaviour of the modified electrode in the presence of different methanol concentrations. It is observed that as methanol concentration increases, the aII peak height increases linearly with methanol concentration up to 0.24 M. It can be assumed that the increment is due to the presence of a diffusion-controlled process that appears to play an important role at low concentrations. While the concentration exceeds from the limit, the rate of whole process seems to be limited by catalytic process in origin and its rate depends on the reaction between methanol and NiOOH species. It is observed that in low methanol concentration, there are two anodic peaks: aI (assigned to oxidation of Ni(OH)2 to NiOOH) and aII (assigned to oxidation of Ni(OH)2 to NiOOH appears only in the presence of methanol). The corresponding electrode reaction involved in the anodic peak aII might be:


It is observed that when methanol concentration increases, peak aII current increases significantly while the peak aI decreases and even disappears when the concentration becomes more than 0.24 M. This result shows that much Ni(OH)2 species is generated through the Equation 5, and subsequently, the peakaII current increases considerably.
3.5. The Effect of Potential Scan Rate
Cyclic voltammograms of the Ni(OH)2NPs/MGCE in the presence of 0.04 M methanol at various potential scan rates were recorded in Fig. 6A. It can be seen from the figure that both methanol and Ni(OH)2 oxidation peaks potentials slightly shift to more positive values with increasing of potential scan rates (except plot (d)), suggesting a kinetic limitation. This finding reveals that the catalytic oxidation of methanol on the modified electrode is not a rapid reaction. The catalytic current for methanol oxidation corresponding to the Equation 6 decreases rapidly with increasing of potential scan rate. Indeed, the time window for catalytic process at higher scan rates becomes very narrow avoiding the facile electron transfer between substrate and catalytic sites. However, the peak currents of Ni(II) oxidation and Ni(III) reduction enhance with increasing of potential scan rate. Therefore, it can be concluded that the Ni(II)/Ni(III) transformation process is much faster than the methanol oxidation. Direct reaction between methanol and Ni(III) produces Ni(II) leading to a little increase in the aI peak current. These results and also plotting current function (anodic peak current (aII) divided by υ1/2) against υ, reveals decreasing curve, confirming the electrocatalytic nature of the process (Fig. 6B).

It is generally agreed that the Ni-based catalysts such as nickel oxides and its complexes in a basic aqueous medium can catalyze the methanol oxidation through an overall four electron process for producing of formate anion:28

3.6. Chronoamperometric Studies
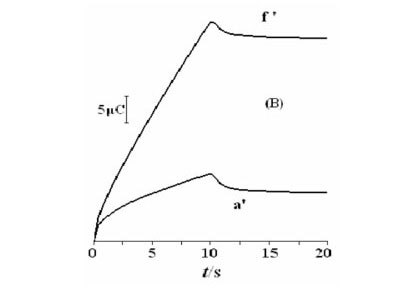
Double potential step chronoamperometry was employed for the investigation of electrochemical processes at the Ni(OH)2NPs/MGCE. Figure 7A shows double-step chrono-amperograms for the modified electrode by setting the working electrode potential at 0.65 V (first step) and 0.25 V (second step) for various methanol concentrations. In the presence of methanol, the charge value associated with the forward chronoamperometry, Qa, is greater than that backward chronoamperometry (Fig. 7B (f')). Chronoamperometry can also be used for evaluation of chemical reaction between methanol and the catalyst layer (catalytic rate constant, k) according to:25


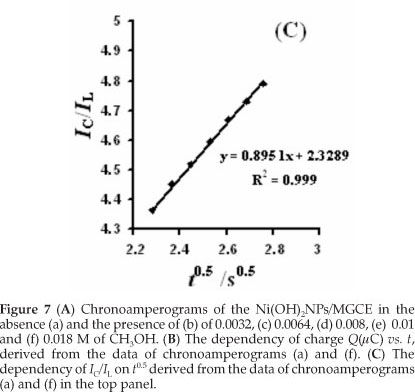
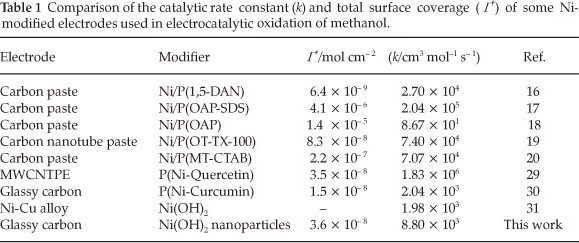
Where IL, IC, k, co and t are the current in the absence and presence of methanol, catalytic rate constant, bulkconcentration of methanol and the elapsed time, respectively. From the slope of the IC/IL vs. t0.5 plot, presented in Fig. 7C, the mean value of k for the concentration range of 0.0032- 0.018 M of methanol is obtained as 8.8 x 103 cm3 mol-1 s-1. This estimated k value is comparable with other k values for similar systems in the literature (Table 1).
3.7. Long-term Stability of the Ni(OH)2NPs/MGCE
Figure 8A shows a chronoamperogram with a large time window of the modified electrode in the presence of methanol. As can be seen, the decrease in current is relatively slow and, when the time is above 200 s, the current reaches a relatively stable value, which is still about 52 % of the initial current. It is obvious that the Ni(OH)2NPs/MGCE exhibits high stability towards methanol oxidation. We also checked long-term stability of the modified electrode by measuring its response for repetitive methanol electrooxidation for first (a) and after 200 cycles (b) of potential cycling (Fig. 8B). It is obvious that after 200 cycles no significant change in peak currents was observed. Also stability of the modified electrode was verified by measuring its response to methanol oxidation after six weeks of storage in the laboratory atmosphere conditions. Such stability seems to be acceptable for practical applications. In comparison with some previous works, it seems clear that Ni(OH)2NPs/MGCE can act as a comparable electrocatalyst in methanol oxidation (Table 2).


4. Conclusion
The electrochemical behaviour of methanol at bare GCE in basic medium is poor and it does not undergo oxidation prior to discharge of the supporting electrolyte. The Ni(OH)2 nano-particles modifying the electrode surface has played a mediated role on heterogeneous catalytic oxidation of methanol in 0.1 M NaOH solution. The oxidation process commences at a potential where the oxidizing Ni(III) species are generated. In the redox system, Ni(OH)2 is firstly oxidized to NiOOH; then, the generated NiOOH is reduced to Ni(OH)2 by methanol. Subsequently, the Ni(OH)2 is converted to NiOOH at more positive potentials, leading to the appearance of a new anodic peak. The methanol oxidation process is diffusion controlled at low concentrations while it is governed by the catalytic reaction between methanol and Ni(III) at higher concentrations. The oxidation process of methanol begins as Ni(III) starts to form on the electrode surface during the positive going potential sweep. The k value indicates that the modified electrode can overcome the kinetic limitation for methanol oxidation by a catalytic process and can decrease the overpotential for its oxidation reaction. The stability of the modified electrode seems to be acceptable for practical applications.
References
1 T. Iwasita, Electrochim. Acta, 2002, 47, 3663. [ Links ]
2 M.A. Abdel Rahim, R.M. Abdel Hameed and M.W. Khalil, J. Power Sources, 2004, 134, 160. [ Links ]
3 A. Verma and S. Basu, J. Power Sources, 2005, 145, 282. [ Links ]
4 H. Heli, M. Jafarian, M.G. Mahjani and F. Gobal, Electrochim. Acta, 2004, 49, 4999. [ Links ]
5 R. Parsons and T. Vandernoot, J. Electroanal. Chem., 1988, 257, 9. [ Links ]
6 K. Nishimura, K. Machida and M. Enyo, J. Electroanal. Chem., 1988, 251, 117. [ Links ]
7 M. Emilia, J.C. Francisco, V. Joseluis and A. Antonio, Electrochim. Acta, 1992, 37, 1883. [ Links ]
8 A.V Tripkovic, N. Marinkovic, K.D. Popovic and R.R. Adzic, Russ J. Electrochem, 1995, 31, 993. [ Links ]
9 M. Chen, Z.B. Wang, Y. Dingand G.P. Yin, Electrochem. Commun., 2008, 10, 443. [ Links ]
10 S.J. Liu, Electrochim. Acta, 2004, 49, 3235. [ Links ]
11 Y. Wu, Y. He, T. Wu, T. Chen, W. Weng and H. Wan, Mater. Lett., 2007, 61, 3174. [ Links ]
12 X. Li,X. Zhang, Z. Liand Y. Qian, Solid State Commun., 2006, 137, 581. [ Links ]
13 H.J. Liu, T.Y. Peng, D. Zhao, K. Dai and Z.H. Peng, Mater. Chem. Phys, 2004, 87, 81. [ Links ]
14 D. Tao and F. Wei, Mater. Lett., 2004, 58, 3226. [ Links ]
15 E.E. Kalu, T.T. Nwoga, V Srinivasan and J.W. Weidner, J. Power Sources, 2001, 92, 163. [ Links ]
16 R. Ojani, J.B. Raoof and S.R. Hosseini, Electrochim. Acta, 2008, 53, 2402. [ Links ]
17 R. Ojani, J.B. Raoof and S. Fathi, Electrochim. Acta, 2009, 54, 2190. [ Links ]
18 R. Ojani, J.B. Raoof and S. Fathi, J. Solid State Electrochem., 2009, 13, 927. [ Links ]
19 J.B. Raoof, R. Ojaniand S.R. Hosseini, J. Power Sources, 2011,196, 1855. [ Links ]
20 J.B. Raoof, M.A. Karimi, S.R. Hosseini and S. Mangelizadeh, J. Electroanal. Chem., 2010, 638, 33. [ Links ]
21 A. Salimi, E. Sharifi, A. Noorbakhsh, S. Soltanian, Biophys. Chem., 2007, 125, 540. [ Links ]
22 S.L. Medway, C.A. Lucas, A. Kowal, R.J. Nichols, D. Johnson, J. Electroanal. Chem, 2006, 587, 172. [ Links ]
23 M. Vukovic, J. Appl. Electrochem, 1994, 24, 878. [ Links ]
24 A.A. El-Shafei, J. Electroanal. Chem, 1999, 471, 89. [ Links ]
25 A.J. Bard and L.R. Faulkner, Electrochemical Methods, Fundamentals and Applications. Wiley and Sons, New York, 2001. [ Links ]
26 J.M. Saveant and E. Vianello, Electrochim. Acta, 1963, 8, 905. [ Links ]
27 M. Fleischmann, K. Korinek and D. Pletcher, J. Electroanal. Chem., 1971, 31, 39. [ Links ]
28 W.S. Cardoso, V.L.N. Dias, W.M. Costa, I.A. Rodrigues, E.P. Marques, A.G. Sousa, J. Boaventura, C.W.B. Bezerra, C. Song, H. Liu, J. Zhang and A.L.B. Marques, J. Appl. Electrochem., 2009, 39, 55. [ Links ]
29 L. Zheng and J.F. Song, J. Solid State Electrochem, 2010, 14, 43. [ Links ]
30 A. Ciszewski, G. Milczarek, B. Lewandowska and K. Krutowska, Electroanalysis, 2003, 15, 518. [ Links ]
31 M. Jafarian, R.B. Moghhaddam, M.G. Mahjani and F. Gobal, J. Appl. Electrochem., 2006, 36, 913. [ Links ]
32 A.N. Golikand, M. Asgari, M. Ghannadi-Maragheh and S. Shahrokhian, J. Electroanal. Chem., 2006, 588, 155. [ Links ]
33 S.J. Liu, Electrochim. Acta, 2004, 49, 3235. [ Links ]
34 A.N. Golikand, S. Shahrokhian, M. Asgari, M. Ghannadi Maragheh, L. Irannejad and A. Khanchi, J. Power Sources, 2005, 144, 21. [ Links ]
Received 29 April 2012
Revised 7 December 2012
Accepted 21 January 2013
* Author for correspondence. E-mail address: j.raoof@umz.ac.ir


{kind=link}