









Servicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
Indicadores

Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkWater SA
versión On-line ISSN 1816-7950
versión impresa ISSN 0378-4738
Water SA vol.36 no.4 Pretoria jul. 2010
Comparison of conventional culture and real-time quantitative PCR using SYBR Green for detection of Legionella pneumophila in water samples
M Fittipaldi; F Codony*; J Morató
Laboratori de Microbiologia Sànitaria y Mediambiental (MSM Lab)- Aquasost, UNESCO Chair in Sustainability, Universitat Politècnica de Catalunya, Edifici Gaia. Pg. Ernest Lluch / Rambla Sant Nebridi. 08222. Terrassa
ABSTRACT
The genus Legionella comprises more than 40 species and 64 serogroups with approximately half of those species associated with human diseases. Legionella pneumophila Serogroup 1 is the most common pathogenic species and is responsible for up to 80% of legionellosis cases in the world. Legionella levels in water are assessed routinely by culture on a selective medium, but its slow growth is a serious drawback, given that at least 10 days are required to obtain results. In an attempt to provide a simple screening method for Legionella pneumophila in water systems samples a real time PCR assay using SYBR Green was developed. A total of 50 samples from cooling towers and hot tap water systems were analysed by DNA amplification using 2 pairs of primers targeting the mip and dot genes. Legionella pneumophila Serogroup 1 (NCTC12821) was used as a reference strain and to evaluate real-time PCR performance. The assays were successful with both primer sets; good and similar amplification efficiencies were achieved. In addition, high sensitivity was obtained; the method proved to allow for the detection of fewer than 10 gene copies per reaction. Results of real-time PCR were compared to conventional analysis based on culture. Although no strong correlation was observed between both methods and consequently real-time PCR could not substitute for the reference method, it represents a powerful screening tool. The inexpensive, sensitive and rapid real-time PCR based in SYBR Green method is of interest in monitoring Legionella pneumophila contamination, especially in environmental samples, and should be economical for large-scale routine tests.
Keywords: Legionella pneumophila, real time PCR, SYBR Green
Introduction
Legionellae are ubiquitous in the natural environment, being present in soils and aquatic ecosystems (Fliermans et al., 1981; Fliermans, 1996). Legionella sometimes survives as an intracellular parasite of amoebae and ciliates (Ratcliff et al., 1998). They are present in process facilities' water systems such as cooling towers, water networks in hospitals, industrial or residential buildings and hydrothermal devices, among others. As a consequence of favourable temperatures, and lack of or poor disinfection, levels of legionellae may be of concern in these systems.
The genus Legionella comprises more than 40 species and 64 serogroups (O'Connell et al., 1996), with approximately half of those species associated with human diseases (Stout and Yu, 1997). Furthermore, Legionella pneumophila Serogroup 1 is the most common pathogenic species and is responsible for up to 80% of legionellosis cases in the world (Doleans et al., 2004). Between 1995 and 2005, a total of 4 056 culture-confirmed cases of Legionnaires' disease from European countries were notified to the European Working Group for Legionella Infections (EWGLI) (Diereden, 2008).
Respiratory infection by Legionella pneumophila (Lp) is mainly attributed to contaminated water aerosols inhalation (Grabow, 1991; Anonymous, 1994), produced by systems such as cooling towers (Rosmini et al., 1984), showers (Mastro et al., 1991), and nebulisers (Blatt et al., 1993). Aspiration of contaminated water aerosols has also been proposed as a possible mechanism of transmission (Yu, 1993; Steinert et al., 1997).
Legionellosis is generally considered to be a preventable illness because controlling or eliminating the bacterium in certain reservoirs will (in theory) prevent disease. This fact has resulted in a number of guidelines and control strategies aimed at reducing the risk of legionellosis in building water systems (Diereden, 2008). A risk assessment and management approach is implemented by the American Society of Heating, Refrigerating and Air Conditioning Engineers (ASHRAE) and the U.K. Health and Safety Executive (Mascone, 2008). Legionella levels in water are assessed routinely by culture on a selective medium like buffered charcoal yeast extract (BCYE) or glycine vancomycin polymyxin cycloheximide agar (GVPC), but its slow growth is a serious drawback, given that it requires at least 10 days to obtain results. Standard culture methods are time consuming and special reagents, culture media and a high degree of technical skill are required in their application (Bartie et al., 2001) because it is difficult to isolate Legionella in waters with high levels of other heterotrophic bacteria. However, until now, standard culture methods have proven to be useful tools. Nevertheless, in the 'hazard analysis critical control point' (HACCP) process in environments or during outbreak investigations this delay is a serious drawback.
In recent years polymerase chain reaction (PCR) methods have become the alternative to detect the presence of Legionella in a few hours, through amplification of specific DNA sequences. The evolution of conventional PCR to real-time PCR has improved this situation even more. The procedure follows the general principle of PCR, but its key feature is that the amplified DNA is quantified as it accumulates in the reaction in real time after each amplification cycle. Two common methods of quantification are the use of fluorescent dyes that intercalate with double-stranded DNA, and modified DNA oligonucleotide probes that fluoresce when hybridised with complementary DNA.
Multiple Legionella detection assays have been published using different target genes (Diederen et al., 2008; Yañez et al., 2005), probe chemistries (Behets et al., 2007; Joly et al., 2006) and real-time thermal cyclers (Stølhaug and Bergh, 2006; Yaradou et al., 2007) but, until now, little has been known about the feasibility of a procedure based on SYBR Green. However, there are some reports which demonstrate that SYBR Green can be used for detection of pathogenic microorganisms (Somogyvari et al., 2007; Kares et al., 2004; O'Mahony and Hill, 2002).
SYBR Green is a non-specific dye which binds to any double-stranded DNA. Therefore, it will not only bind to PCR products but to non-specific products that have been amplified, as well as primer dimers. Due to this non-specificity, SYBR Green requires more time for optimisation than protocols based on primers and probes. Nevertheless, when SYBR Green is used as the fluorescent dye, a subsequent melting curve analysis of PCR products generates a specific profile (depicting the fluorescence change rate over time as a function of temperature for each sample), which can be used to determine the success of the PCR reaction (O'Mahony and Hill, 2002). Furthermore, the use of master mixes based on SYBR Green chemistries is simple, fast and inexpensive.
The objective of this study was to develop a simple, screening real-time PCR assay for Legionella pneumophila in water systems samples, using SYBR Green-based detection and two pairs of primers targeting the mip and dot genes. These primers were previously published as part of a set of primers-probes (Hayden et al., 2001; Yañez et al., 2005). Results of real-time PCR were compared with conventional analysis based on culture.
Materials and methods
Water samples and culture
A total of 50 samples were analysed. All samples were from urban areas around Barcelona city (north-east Spain), with two different origins: cooling tower (20 samples) or hot tap water from big buildings (30 samples). Water samples were collected in Pyrex bottles and the quantification of Lp by culture was performed according to international standard ISO 11731:1998 (ISO) using culture media (GVPC) and reagents from OXOID. The quantification limit was 1 x 102 CFU/ℓ.
Sample concentration and DNA extraction
A litre of water for each sample was concentrated by membrane filtration using a nylon membrane (0.45 µm pore diameter, Millipore). Cells were re-suspended in 10 mℓ of saline solution by vigorous vortexing for 60 s with 5 glass beads (5 mm diameter) and sonication for 3 min in an ultrasound water bath (Selecta 40W power, 40 kHz ultrasound frequency). The cell suspension was again concentrated to 2 mℓ and then to 150 µℓ by centrifugation (14 000 revolutions/min, 5 min) using a MiniSpin centrifuge (Eppendorf, Hamburg, Germany). DNA was extracted with DNeasy Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions.
Real-time PCR optimisation
The procedure was set up using previously reported primers targeting dot (Yañez et al., 2005) and mip genes (Hayden et al., 2001). Three key points of real-time PCR reactions were considered for method adaptation and optimisation: annealing temperature and primers and MgCl2 concentrations. Optimisation of each parameter was performed by modifying one of them while keeping the rest fixed at a given value (one-factor-at-a-time, OFAT). The sequence used was the same described previously (temperature, primer concentration and MgCl2 concentration). At each step, the optimum value of each parameter was selected using as criteria the highest annealing temperature and the lowest threshold cycle (Ct); under those conditions dimerization is minimised. Assays were performed in triplicate in all cases. Real-time PCR conditions evaluated with the selected primers considered annealing temperatures of 55, 56, 57, 58, 59, 60, 61 and 62ºC; primer concentrations of 0.50, 0.75, 1.00 and 1.50 µM and MgCl2 concentrations of 1, 2, 3, 4 and 5 mM.
Evaluation of optimal annealing temperature and primer concentration was carried out using a Quantitect SYBR Green PCR kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. Determination of MgCl2 concentration was performed using a Lightcycler 1.5 (Roche Molecular Diagnostic, Manheim, Germany) in combination with FastStart DNA masters SYBR Green 1 kit (Roche, Germany), according to the manufacturer's instructions. Contribution of MgCl2 to the reaction efficiency, at optimum temperature and primer concentrations, was evaluated using a different mix, because this commercial mix does not contain MgCl2 and the level has to be fixed by the user; other commercial mixes, such as Quantitect SYBR Green PCR kit (Qiagen, Valencia, CA), usually contain MgCl2 in optimum concentration for most purposes.
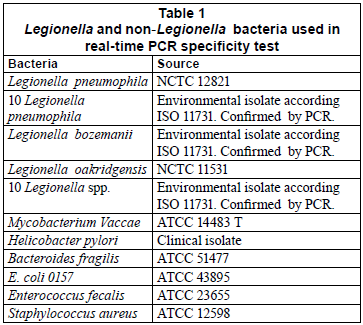
Primer specificity was re-evaluated in order to ensure their good performance in the absence of an internal control. First, a GenBank query was performed. Second, the specificity of the real-time PCR assay was investigated using DNA extracts of Legionella pneumophila, Legionella species and non-Legionella bacteria (Table 1). After culture on their appropriate medium for 24 h, bacterial cells were harvested and suspended in saline solution. After adjusting the concentration of cell suspension at 600 nm, it was 10-fold diluted and a 200 µℓ aliquot of the dilution related to 106 colony-forming units (CFU/mℓ) was extracted with the DNeasy Tissue Kit (Qiagen, Valencia, CA).
Legionella pneumophila DNA standard
The DNA used for reaction validation and as reference for Lp quantification in water samples was prepared according to AFNOR XP T90-471. Lp Serogroup 1 (NCTC12821) was used as a reference strain and to evaluate real-time PCR performance.
A standard DNA curve was established using a 4-day culture. DNA was obtained with the DNeasy Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The DNA amount was determined by measuring florescence at 530 nm using the Nucleic Acid Quantification method from Lightcycler 1.5 (Roche). SYBR Green and DNA standard (Maize GMO Standard for NK 603, Fluka Biochemika, Sigma) were used to quantify DNA. DNA concentration was about 30(±0.5) ng/µℓ, as the genome of Legionella pneumophila is 4.3 fg (according to AFNOR XP90-471), and the number of DNA genomic units (GU) of Lp theoretically contained in the extract was evaluated to be 6.05 106 GU/µℓ. Consequently, serial logarithmic dilutions in PCR water, from 101 to 106 GU per reaction, were performed. Amplification efficiency was estimated by means of the slope calculation method from a calibration dilution curve (Rasmussen, 2001). In each case the standard curve was performed by duplicate.
In order to compare GU and CFU values for pure culture, Legionella pneumophila Serogroup 1 was cultured in Legionella selective medium GVPC (Oxoid) for 4 days at 37ºC. Once the culture was ready, a bacterial suspension was prepared transferring single colonies into sterile saline solution and adjusting the optical density (measured at 600 nm) to 0.2 using a Pharmacia Biotech Novaspec® II spectrophotometer. Serial dilutions were prepared. Each solution was tested by culture and real-time PCR for mip and dot genes using Lightcycler 1.5 (Roche). These assays were performed in duplicate and the corresponding mean values were calculated. Dissociation curves were also recorded after each run.
Real-time PCR assays
Water sample analysis was performed on a Lightcycler 1.5 (Roche). The reaction mixtures for both primers were composed of 9 µℓ SYBRGreen (Quantitect SYBR Green PCR kit, Quiagen, Valencia, CA), 0.4 U of Uracil-DNA-glycosylase (UDG, New England Biolabs, UK), 9 µℓ of sample and 0.5 µM of mip primers or 0.75 µM of dot primers respectively. These concentrations were the results of the real-time PCR optimisation assay.
The experimental protocol was as follows: 2 min at 50ºC to allow UDG to break down the possible contaminating amplicons, 15 min at 95ºC for Taq polymerase activation, 45 cycles (94ºC for 15 s, 59ºC for 30 s and 72ºC for 30 s) for DNA amplification and finally a melting temperature ramp from 65ºC to 95ºC at 0.1ºC/s.
For each assay, Ct was determined in order to quantify each DNA product. Quantification was performed by including 1 or 2 external standards, theoretically containing 4.5 104 GU, in each set of PCR experiments. The GU number of each sample was determined by comparison to each standard. Each sample was tested in duplicate and the mean value was used for statistical analysis. A negative control (water, PCR grade) was included in all assays. Melting curve analysis was done for all assays to evaluate positive and negative results.
Statistical analyses
In order to compare the equivalence between PCR and conventional microbiology, qualitative results for ISO/TR 13843:2000 is used as a reference. Statistical analysis to compare culture and PCR results was performed using Microsoft Excel and correlation was established using Pearson's coefficient. A Chisquare test was used to examine whether the results of 2 methods (culture and real-time PCR) were independent or not. This test uses a null hypothesis that the results achieved by culture and real-time PCR are independent. Consequently, a false null hypothesis means that culture and real-time PCR are dependent. In this case Yates' correction was used due to the reduced sample size, given that use of the chi-square distribution can introduce some bias to the calculations by making the value of statistical chi-square greater (Fleiss et al., 2003).
Results
Real-time PCR optimisation
We adapted and validated a real-time PCR procedure based on SYBR Green with 2 pairs of primers that targeted the mip and dot genes. We found that the optimal annealing temperature for the primers was 59ºC. The optimal concentrations were 0.5 µM for the mip primer set and 0.75 µM for the dot primer set. The optimisation of the MgCl2 concentration in the master mix indicated that between 3 or 4 mM was an optimal concentration for both primer sets. The Quantitect SYBR Green PCR kit (Qiagen, Valencia, CA) provided a 4 mM MgCl2 concentration.
The specificity of the SYBR Green assay was tested on a panel of Legionella and non-Legionella bacteria. The results showed positive signals (mean Ct values of 18.22) for all Legionella pneumophila bacteria (the reference strain and the environmental isolates). The amplification of the other Legionella species and non-Legionella bacteria produced only faint signals (Ct values higher than 35) and showed negative signals in the melting curve and agarose gel analyses.
Standard curve
A linear regression analysis was performed by plotting the Ct values against the logarithm of the copy number of each gene target. We found that the experimental points aligned in a straight line with correlation coefficients (R) of -0.994 (R2=0.9885) and -0.996 (R2=0.9923) for mip and dot, respectively.
For mip gene detection, the equation from the regression curve was: Ct = -3.4135 log [GU] + 39.356. The slope of -3.4135 corresponded to an amplification efficiency of 98.1%. The assay showed a sensitivity of 9 GU per reaction (mean Ct = 35.3; Fig. 1A). The quantification limit was estimated to be 9.60 102 GU/ℓ of sample. The coefficient of variation ranged from 1.7% to 2.8%, depending on the concentration of the DNA sample.
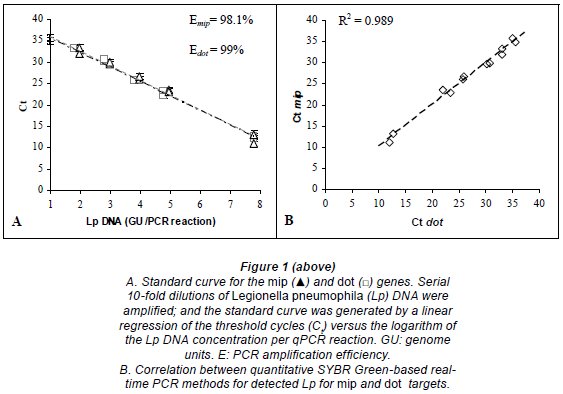
For dot gene detection, the equation from the regression curve was: Ct = -3.3698 log [GU] + 38.752. The slope of -3.3698 corresponded to an amplification efficiency of 99%. The assay showed a sensitivity of approximately 9 GU per reaction (mean Ct = 35.2; Fig. 1A). The quantification limit was estimated to be 9.58 102 GU/ℓof sample. The coefficient of variation ranged from 0.2% to 3.5%, depending on the concentration of the DNA template.
The mip and dot primers gave similar results for Legionella pneumophila quantification by real-time PCR. A comparison of the number of cycles required for detectable amplification with each primer showed a linear relationship from 6 to 6·106 GU/reaction (R2 = 0.989; Fig. 1B).
Comparison between results from quantitative PCR and Lp cultures
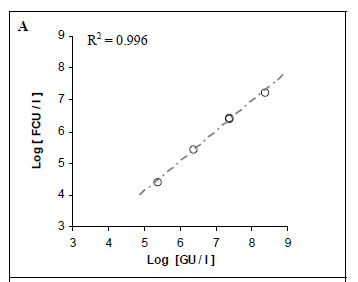
Pure suspensions of Lp were analysed by both real-time PCR and culture methods. We aimed to compare the quantitative results and determine whether they were correlated. A strong correlation was found between the positive, quantifiable results produced by the 2 real-time PCR methods (R2 = 0.996; Fig. 2A).
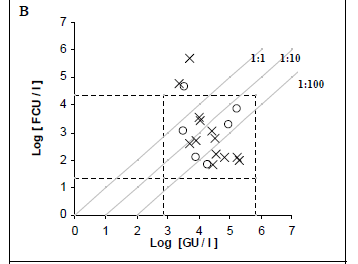
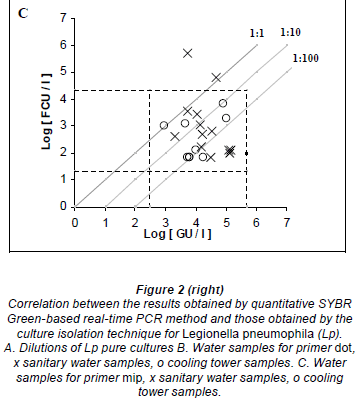
Fifty purified samples were analysed by culture and real-time PCR methods. Among these samples, 22 (44%) were culture positive and the remaining 28 (56%) were culture negative. The real-time PCR method indicated that, among the 22 culture-positive samples, 19 (86.4%) and 21 (95.4%) were positive for the dot and mip genes, respectively. Moreover, the real-time PCR showed that, among the 28 culture-negative samples, 7 (25%) and 10 (35.7%) were positive for mip and dot, respectively. The real-time PCR results were an average of 2.07 log higher than culture results for detecting the mip gene, with a margin of error of 0.3368 and a confidence interval (CI) of 95%. Similarly, the real time PCR results were an average of 1.77 log higher than culture results for detecting the dot gene, with a margin of error of 0.4306 and a CI of 95% (Figs. 2C and 2B).
The melting curves and gel electrophoresis for both primer sets displayed single peaks and bands, respectively. In contrast, no peak or band was displayed in the negative controls. This indicated that these primer sets were specific for Legionella pneumophila (Fig. 3).
To compare the culture and real-time PCR methodologies, a linear regression was performed for all samples that had quantifiable amounts of Lp by both methods. A statistical analysis did not reveal any correlation between the two methods (R2 = 0.137 for mip and R2 = 0.234 for dot). However, there was a general association between a large number of colonies detected by culture and a high number of Lp genome units detected by real-time PCR.
Thirty hot tap-water samples were analysed. Among these samples, 14 (46.7%) were positive and 16 (53.3%) were negative by culture. Among the 14 culture-positive samples, 13 (92.8%) were positive for mip and dot by real-time PCR, and only one was negative by PCR. Real-time PCR results were, on average, 2.03 log higher than culture results for mip PCR, and 2.32 log higher than culture results for dot PCR. Among the 16 culture-negative samples, 5 (31.2%) and 6 (37.5%) were positive for mip and dot, respectively, by real- time PCR.
Twenty cooling tower samples were analysed. Among these, 8 (40%) were positive and 12 (60%) were negative by culture. Among the 8 culture-positive samples, 8 (100%) and 6 (75%) were positive for mip and dot, respectively, by real-time PCR. Real-time PCR results were, on average, 2.65 log higher than culture results for detecting the mip gene, and 2.10 log higher than culture results for detecting the dot gene. Among the 12 culture-negative samples, 2 (16.7%) were positive for mip, 3 (25%) were positive but non-quantifiable, and 1 (8.3%) was positive for dot by real- time PCR.
To compare the culture and real-time PCR methodologies, a linear regression was performed for all the samples that had quantifiable amounts of Lp by both methods. One regression was performed for the samples from each origin. The statistical analyses on both the sanitary and cooling tower water results did not reveal any strong correlations between the culture and real-time PCR methods. But, in general, the correlation for sanitary samples tended to be stronger than that observed for cooling tower samples.
Chi-Square Test (X2)
Two-by-two contingency tables for each primer were constructed (Table 2). The X2 was calculated for each table, considering 1 degree of freedom for both tables, and a p-value < 0.01 (α = 0.01). According to the chi-square distribution, for 1 degree of freedom and α = 0.01, the critical value is 6.63. Thus, when a X2 is greater than 6.69, the difference is significant, and the null hypothesis is rejected.
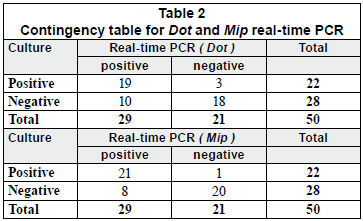
In this study, the X2 were found to be 22.62 and 12.97 for mip and dot contingency tables, respectively. These results clearly showed that the culture and real-time PCR methods were correlated.
Discussion
In this report, we described a simple, sensitive, reliable realtime PCR method for detecting Legionella pneumophila in water samples. We developed and optimised a real-time quantitative PCR assay based on SYBR Green and 2 pairs of primers that targeted the mip and dot genes. Three key features of PCR reactions were considered for optimisation: the annealing temperature, the primer concentration and the MgCl2 concentration. Both primers were found to have the same high annealing temperature. A high annealing temperature is desirable because it enhances the specificity of the dye binding to the DNA and avoids inhibition and primer-dimer formation.
In order to quantify the number of copies of Lp in the water samples, we constructed a standard curve for each gene target. High amplification efficiencies were achieved for both the mip and dot primers (1.96 and 1.98, respectively). The amplification curves were highly correlated. Furthermore, a linear relationship was observed between the real-time PCR quantification of Legionella pneumophila by mip and dot primers; thus, both primers would be useful in a specific, fast, economical screening method for detecting Lp in water samples.
When we worked with pure cultures, we observed a strong linearity between the results achieved with real-time PCR with SYBR Green and those obtained with conventional culture techniques. Joly et al. (2006) and Yañez et al. (2005) came to a similar conclusion. However, when we compared the conventional cultures and SYBR Green-based real-time PCR methods with different environmental water samples (hot sanitary and cooling tower samples), we found weak correlation coefficients. Nevertheless, a tendency was observed towards higher correlation coefficients for hot sanitary samples than for cooling tower samples, particularly in detecting the dot gene. Several studies have reported similar results with real-time PCR (Morio et al., 2008; Wellinghausen et al., 2001; Yaradou et al., 2007; Joly et al., 2006).
Although no strong correlation was observed in the comparisons of the 2 methods, the statistical analysis showed that a large number of colonies detected in culture was generally associated with a high number of genome units of Legionella pneumophila detected by real-time PCR. We observed that SYBR Green-based real-time PCR results were, on average, 1 log and 1.9 log higher than conventional culture results, for pure culture samples and both cooling tower and sanitary samples, respectively. Similar results have been observed in other studies (Wellinghausen et al., 2001; Yañez et al., 2005; Morio et al., 2008). These differences may arise from different causes. Real-time PCR can detect all cells, both viable (culturable and non-culturable) and non-viable; by contrast, culture methods can only detect viable, culturable cells (Hussong et al., 1987). The results from these methods are expressed in different units - the CFU is the unit used in cultures and the GU is used in real-time PCR. To our knowledge, no methods have been established for deriving equivalent units for comparisons (Stølhaug and Bergh, 2006). However, in our case an equivalence could be reached by comparing real-time PCR and culture methods using pure Legionella pneumophila suspensions. Wellinghausen et al. (2001) showed that DNA extraction enabled the detection of legionellae in free-living amoeba, but culturing methods could not detect amoeba. Furthermore, standard culture techniques based on ISO 11731 have numerous limitations (Behets et al., 2007) and several factors complicate the interpretation of plate-counting results (Devos et al., 2005). Cultures can be fastidious due to various factors; for example: the legionellae growth requirements necessitate prolonged incubation periods; the legionellae are difficult to isolate in samples contaminated with high levels of other microbiota, particularly environmental samples; the pre-treatment by acid or heating can lead to underestimates of the number of viable legionellae; viable, but non-culturable bacteria cannot be detected (Catalan et al., 1997; Wellinghausen et al., 2001; Yañez et al., 2005); and Legionella spp. are present at low densities in environmental samples.
Although diagnostic methods have improved since Lp was first described in 1976, no test is currently available that can diagnose Lp in a timely fashion with a high degree of sensitivity and specificity (Diederen, 2008). Real-time PCR methods offer the benefit of speed over traditional culturing methods, and allow earlier disinfection of water systems that contain high numbers of Legionella pneumophila bacteria (Behets et al., 2007). The major disadvantage of real-time PCR lies in its inability to differentiate between viable and non-viable cells. This is important in monitoring Legionella contamination levels in environmental samples. However, several studies have reported the use of nucleic acid-binding dyes as an attractive alternative for selectively detecting and enumerating viable bacteria (Chang et al., 2009; Delgado-Viscogliosi et al., 2009).
Occasionally, SYBR Green-based detection strategies in real-time PCR analyses have been criticised for their impracticality, due to the lack of dye specificity for binding to DNA (Hein et al., 2001). There is a common misconception that adding an oligoprobe to a reaction will automatically make the reaction more sensitive. However, specific and non-specific fluorogenic chemistries are able to detect amplicons with equal sensitivity (Newby et al., 2003; Fernández et al., 2006). The presence of primer-dimers or non-specific products can sometimes be detected in the melting curve analysis.
Conclusions
To our knowledge, this is the first report on the use of real-time PCR in combination with SYBR Green for the quantification and identification of Legionella pneumophila in pure cultures and environmental samples. We achieved good results in detecting Legionella pneumophila with an inexpensive, SYBR Green-based, quantitative, real-time PCR method. Furthermore, the results achieved were comparable to those obtained with real-time assays that employed expensive fluorescence resonance energy transfer probes (FRET). Non-specific chemical methods like SYBR Green are relatively inexpensive, do not require additional oligonucleotide design or chemical conjugation, and are minimally affected by small changes in template sequences. In contrast, small changes in template sequences can abolish the hybridisation of an oligoprobe, even with primers that have previously amplified the template successfully (Komurian-Pradel et al., 2001). Therefore, the SYBR Green method appears to be a suitable alternative for monitoring Lp contamination, particularly in environmental samples, and should be economically feasible for large-scale routine testing. More studies that investigate real-time PCR assays for Legionella pneumophila detection are necessary to stimulate broader use of standard PCR methods. The detection and quantification of Legionella by real-time PCR could play key roles, both during an outbreak investigation and in the context of a health risk management programme.
Acknowledgements
We are most grateful to the Agència de Gestió d'Ajuts Universitaris i of Recerca for the financial support received. Aigües de Barcelona Company is gratefully acknowledged for supplying Legionella pneumophila and Legionella spp. bacteria. The authors would also like to acknowledge the advice and support kindly given by Veronica Rajal.
References
AFNOR XP T90-471 (2006) Détection et quantification des Legionella et/ou Legionella pneumophila par concentration et amplification génique par Reaction de polymérisation en chaîne (PCR). Normalization Française. [ Links ]
ANONYMOUS (1994) Legionnaires' disease associated with cooling towers - Massachusetts, Michigan and Rhode Island. Morb. Mortal. Wkly Rep. 43 491-499. [ Links ]
BARTIE C, VENTER SN and NEL LH (2001) Evaluation of detection methods for Legionella species using seeded water samples. Water Sa 27 523-527. [ Links ]
BLATT SP, PARKINSON MD, PACE E, HOFFMAN P, DOLAN D, LAUDERDALE P, ZAJAC RA and MELCHER GP (1993) Nosocomial Legionnaires' disease: aspiration as a primary mode of disease acquisition. am. J. Med. 95 16-22. [ Links ]
BEHETS J, DECLERCK P, DELAEDT Y, CREEMERS B and OLLEVIER F (2007) Development and evaluation of a Taqman duplex real-time PCR quantification method for reliable enumeration of Legionella pneumophila in water samples. J. Microbiol. Methods 68 137-144. [ Links ]
CATALAN V, GARCIA MORENO C, VILA MJ and APRAIZ D (1997) Detection of Legionella pneumophila in wastewater by nested polymerase chain reaction. Res. Microbiol. 148 71-78. [ Links ]
CHANG B, SUGIYAMA K, TAGURI T, AMEMURA-MAEKAWA J, KURA F and WATANABE H (2009) Specific detection of viable Legionella cells by combined use of photoactivated ethidium monoazide and PCR/real-time PCR. appl. Environ. Microbiol. 75 147-153. [ Links ]
DELGADO-VISCOGLIOSI P, SOLIGNAC L, DELATTRE JM (2009) Viability PCR, a Culture-Independent Method for Rapid and Selective Quantification of Viable Legionella pneumophila Cells in Environmental Water Samples. appl. Environ. Microbiol. 75 3502-3512. [ Links ]
DEVOS L, CLYMANS K, BOON N and VERSTRAETE W (2005) Evaluation of nested PCR assays for the detection of Legionella pneumophila in a wide range of aquatic samples. J. appl. Microbiol. 99 916-925. [ Links ]
DIEDEREN BMW (2008) Legionella spp. and Legionnaires' disease. J. Infect. 56 1-12. [ Links ]
DIEDEREN B, KLUYTMANS JAJW, VANDENBROUCKE-GRAULS CM and PEETERS MF (2008) Utility of Real-Time PCR for Diagnosis of Legionnaires'Disease in Routine Clinical Practice. J. clin. Microbiol. 46 671-677. [ Links ]
DOLEANS AH, AURELL M, REYROLLE G, LINA J, FRENEY F, VANDENESH J E and JARRAUD S (2004) Clinical and environmental distributions of Legionella strains in France are different. J. clin. Microbiol. 42 458-460. [ Links ] In: Yaradou DF, Hallier-Soulier S, Moreau S, Pty F, Hillion Y, Reyrolle M, André J, Festoc G, Delabre K, Vandenesch F, Etienne J and Jarraud S (2007) Integrated realtime PCR for detection and monitoring of Legionella pneumophila in water systems. appl. Environ. Microbiol. 73 1452-1456. [ Links ]
FERNÁNDEZ F, GUTIÉRREZ J, SORLÓZANO A, ROMERO JM, SOTO MJ and RUIZ-CABELLO F (2006) Comparison of the SYBR Green and the hybridization probe format for real-time PCR detection of HHV-6. Microbiol. Res. 161 158-163. [ Links ]
FLEISS JL, LEVIN B and PAIK MC (2003) Statistical Methods for Rates and proportions (3rd edn.). John Wiley & Sons, Hoboken, NJ. [ Links ]
FLIERMANS CB, CHERRY WB, ORRISON LH, SMITH SJ, TISON DL and POPE DH (1981) Ecological distribution of Legionella pneumophila. appl. Environ. Microbiol. 41 9-16. [ Links ]
FLIERMANS CB (1996) Ecology of Legionella: from data to knowledge, a little wisdom. Microb. Ecol. 32 203-228. [ Links ]
GRABOW WOK (1991) New trends in infections associated with swimming-pools. Water Sa 17 173-177. [ Links ]
HAYDEN RT, UHL JR, QIAN X, HOPKINS MK, AUBRY MC, LIMPER AH, LLOYD RV and COCKERILL FR (2001) Direct detection of Legionella species from bronchoalveolar lavage and open lung biopsy specimens: Comparison of LightCycler PCR, in situ hybridization, direct fluorescence antigen detection, and culture. J. clin. Microbiol. 39 2618-2626. [ Links ]
HEIN I, LEHNER A, RIECK P, KLEIN K, BRANDL E and WAGNER M (2001) Comparison of different approaches to quantify Staphylococcus aureus cells by real-time quantitative PCR and application of this technique for examination of cheese. appl. Environ. Microbiol. 67 3122-3126. [ Links ] In: O'Mahony J and Hill C (2002) A real time PCR assay for the detection and quantitation of Mycobacterium avium subsp. paratuberculosis using SYBR Green and the LightCycler. J. Microbiol. Methods 51 283-293. [ Links ]
HUSSONG D, COLWELL RR, O'BRIEN MO, WEISS E, PEARSON AD, WEINER RM and BURGE WD (1987) Viable Legionella pneumophila not detectable by culture on agar media. Biotechnol. 5 947-950. [ Links ]
ISO (2000) Water quality - guidance on Validation of Microbiological Methods. ISO/TR 13843:2000. International Organisation for Standardization, Geneva. [ Links ]
JOLY P, FALCONNET PA, ANDRÉ J, WEILL N, REYROLLE M, VANDENESCH F, MAURIN M, ETIENNE J and JARRAUD S (2006) Quantitative real-time Legionella PCR for environmental water samples: data interpretation. appl. Environ. Microbiol. 72 2801-2808. [ Links ]
KARES S, LÖNNROT M, UVORINEN P, OIKARINEN S, TAURIANEN S and HYÖTY H (2004) Real time PCR for rapid diagnosis of entero-and rhinovirus infections using LightCycler. J. clin. Virol. 29 99-104. [ Links ]
KOMURIAN-PRADEL F, PARANHOS-BACCALÀ G, SODOYER M, CHEVALLIER P, MANDRAND B, LOTTEAU V and ANDRT P (2001) Quantitation of HCV RNA using real-time PCR and fluorimetry. J. Virol. Methods 95 111-119. [ Links ]
MASCONE CF (2008) Legionella: an invisible risk. chem. Eng. prog. 104 6-10. [ Links ]
MASTRO TD, FIELDS BS, BREIMAN RF, CAMPBELL J, PLIKAY-TIS BD and SPIKA JS (1991) Nosocomial Legionnaires' disease and use of medication nebulizers. J. Infect. Dis. 163 667-671. [ Links ]
MORIO F, CORVEC S, CAROFF N, LE GALLOU F, DRUGEON H and REYNAUD A (2008) Real-time PCR assay for the detection and quantification of Legionella pneumophila in environmental water samples: Utility for daily practice. Int. J. Hyg. Environ. Health 211 403-411. [ Links ]
NEWBY DT, HADFIELD TL and ROBERTO FF (2003) Real-time PCR detection of Brucella abortus: a comparative study of SYBR Green, I., 5'-Exonuclease, and hybridization probe assays. appl. Environ. Microbiol. 69 4753-4759. [ Links ]
O'CONNELL W A, DHAND L and CIANCIOTTO NP (1996) Infection of macrophage-like cells by Legionella species that have not been associated with disease. Infect. Immun. 64 4381-4384. [ Links ]
O'MAHONY J and HILL C (2002) A real-time PCR assay for the detection and quantitation of Mycobacterium avium subsp. paratuberculosis using SYBR Green and the LightCycler. J. Microbiol. Methods 51 283-293. [ Links ]
RASMUSSEN R (2001) Quantification on the LightCycler. In: Meuer S, Witwe C and Nakagawara K (eds.) Rapid cycler RealTime pcR, Methods and applications. Springer, Berlin. 21-34. [ Links ]
RATCLIFF RM, LANSER JA, MANNING PA and HEUZEN-ROEDER MW (1998) Sequence-based classification scheme for the genus Legionella targeting the mip gene. J. clin. Microbiol. 36 1560-1567. [ Links ]
ROSMINI F, CASTELLANI-PASTORIS M, MAZZOTTI MF, FORASTIERE F, GAVAZZONI A, GRECO D, RUCKDESCHEL G, TARTAGNI E, ZAMPIERI A and BAINE WB (1984) Febrile illness in successive cohorts of tourists at a hotel on the Italian Adriatic coast: evidence for a persistent focus of Legionella infection. am. J. Epidemiol. 119 124-134. [ Links ]
SOMOGYVARI F, DOCZI L, SERLY J, AHMAD S and NAGY E (2007) Rapid discrimination between candida albicans and candida dubliniensis by using real-time polymerase chain reaction. Diagn. Microbiol. Infect. Dis. 58 367-369. [ Links ]
STEINERT M, EMODY L, AMANN R and HACKER J (1997) Resuscitation of viable but non-culturable Legionella pneumophila Philadelphia JR32 by acanthamoeba castellanii. appl. Environ. Microbiol. 63 2047-2053. [ Links ]
STOUT JE and YU VL (1997) Legionellosis. N. Engl. J. Med. 337 682-687. [ Links ]
STØLHAUG A and BERGH K (2006) Identification and differentiation of Legionella pneumophila and Legionella spp. with real-time PCR targeting the 16S rRNA gene and species identification by mip sequencing. appl. Environ. Microbiol. 72 6394-6398. [ Links ]
WELLINGHAUSEN N, FROST C and MARRE R (2001) Detection of legionellae in hospital in water samples by quantitative real-time LightCycler PCR. appl. Environ. Microbiol. 67 2047-2053. [ Links ]
YAÑEZ MA, CARRASCO-SERRANO C, BARBERÁ VM and CATALÁN V (2005) Quantitative detection of Legionella pneumophila in water samples by immunomagnetic purification and real-time PCR amplification of the dota gene. appl. Environ. Microbiol. 71 3433-3441. [ Links ]
YARADOU DF, HALLIER-SOULIER S, MOREAU S, POTY F, HILLION Y, REYROLLE M, ANDRÉ J, FESTOC G, DELABRE K, VANDENESCH JE and JARRAUD S (2007) Integrated realtime PCR for detection and monitoring of Legionella pneumophila in water systems. appl. Environ. Microbiol. 73 1452-1456. [ Links ]
YU VL (1993) Could aspiration be the major mode of transmission for Legionella? am. J. Med. 95 13-15. [ Links ]
Received 28 October 2009; accepted in revised form 31 May 2010.
* To whom all correspondence should be addressed. +34 93-7398566; fax: +34 93-7398238; e-mail: codony@oo.upc.edu

{kind=link}